![[单细胞转录组分析系列教程 1] 怎么做单细胞10X数据读取和预处理?](/media/blog/covers/cover_20250819.jpg)
大家晚上好,在接下来的一个月,给大家更新一期单细胞转录组的分析流程。
在单细胞转录组分析中,数据读取和预处理是整个分析流程的第一步,也是最关键的一步。10X Genomics作为当前主流的单细胞测序平台,产生的数据格式有其特定的结构和要求。很多初学者在这一步就遇到困难,不知道如何正确读取和整理多样本的10X数据。本文将详细介绍如何高效地读取和预处理10X单细胞数据。
# 加载必要的包
library(Seurat)
library(ggplot2)
library(dplyr)
library(stringr)
# 创建输出目录
if (!dir.exists("picture")) {
dir.create("picture")
}
# 设置数据路径
data_path <- "../GSE231559_RAW/"
# 检查数据文件
fs <- list.files(data_path, '^GSM')
print("原始文件列表:")
print(fs)
10X数据的标准格式包含三个核心文件:barcodes.tsv.gz(细胞条形码)、features.tsv.gz(基因特征信息)和matrix.mtx.gz(表达矩阵)。在处理多样本数据时,我们首先需要识别和整理这些文件的结构。从GEO数据库下载的数据通常将所有文件放在同一目录下,文件名包含样本ID信息。
通过提取文件名中的样本ID,我们可以自动识别有多少个样本,并为每个样本创建独立的文件夹。这种文件组织方式不仅便于后续的数据读取,也有利于数据的管理和追溯。
# 提取样本ID
samples <- stringr::str_split(fs, '_', simplify = TRUE)[, 1]
unique_samples <- unique(samples)
print(paste("检测到", length(unique_samples), "个样本:", paste(unique_samples, collapse = ", ")))
# 批量整理文件结构
lapply(unique_samples, function(x) {
y <- fs[grepl(x, fs)]
folder <- file.path(data_path, str_split(y[1], '_', simplify = TRUE)[, 1])
if (!dir.exists(folder)) {
dir.create(folder, recursive = TRUE)
# 移动并重命名文件
file.rename(file.path(data_path, y[grep("barcodes", y)]),
file.path(folder, "barcodes.tsv.gz"))
file.rename(file.path(data_path, y[grep("features", y)]),
file.path(folder, "features.tsv.gz"))
file.rename(file.path(data_path, y[grep("matrix", y)]),
file.path(folder, "matrix.mtx.gz"))
}
})
文件整理完成后,我们需要读取样本的分组信息。通常这些信息存储在单独的临床信息文件中,包含样本ID和对应的实验条件或疾病状态。这些元数据对于后续的差异分析和生物学解释至关重要。
在本例中,我们使用的是肝癌相关的单细胞数据,包含3个正常肝脏样本和3个肝脏肿瘤样本。这种配对的实验设计是单细胞研究中的常见模式,能够有效地识别疾病相关的细胞类型变化。
# 读取临床信息
clinical <- read.table("../clinical.tsv", header = TRUE, sep = "\t")
print("临床信息:")
print(clinical)
# 逐个读取样本数据并创建Seurat对象
seurat_list <- list()
for (sample_id in names(sample_dirs)) {
# 读取单个样本数据
counts_i <- Read10X(data.dir = sample_dirs[sample_id])
# 创建Seurat对象
seurat_i <- CreateSeuratObject(counts_i,
min.cells = 0,
min.features = 0,
project = sample_id)
# 添加到列表
seurat_list[[sample_id]] <- seurat_i
cat("样本", sample_id, "读取完成,细胞数:", ncol(seurat_i), ",基因数:", nrow(seurat_i), "\n")
}
# 合并所有样本
scRNA <- merge(seurat_list[[1]], y = seurat_list[-1],
add.cell.ids = names(seurat_list))
数据读取过程中有几个关键点需要注意。首先,不同样本的基因数量可能不完全相同,直接合并可能会出错,所以采用逐个读取再合并的策略更加稳妥。其次,CreateSeuratObject函数中的min.cells和min.features参数控制基因和细胞的过滤阈值,在初始读取阶段建议设为0,将过滤步骤留到专门的质控环节。
merge函数中的add.cell.ids参数非常重要,它为每个细胞添加样本前缀,确保来自不同样本的细胞具有唯一标识。这对于后续的批次效应分析和样本间比较至关重要。
# 添加样本分组信息
scRNA$group <- ifelse(scRNA$orig.ident %in% c("GSM7290760", "GSM7290764", "GSM7290765"),
"Normal", "Tumor")
# 生成样本统计图
sample_stats <- table(scRNA$orig.ident, scRNA$group)
print("样本细胞数统计:")
print(sample_stats)
# 图1: 样本细胞数柱状图
p1 <- scRNA@meta.data %>%
count(orig.ident, group) %>%
ggplot(aes(x = orig.ident, y = n, fill = group)) +
geom_col(position = "dodge", alpha = 0.8) +
scale_fill_manual(values = c("Normal" = "#4A90E2", "Tumor" = "#E24A4A")) +
labs(title = "Cell Count per Sample", x = "Sample ID", y = "Cell Number", fill = "Group") +
theme_minimal() +
theme(
panel.grid = element_blank(),
axis.text.x = element_text(angle = 45, hjust = 1, size = 10),
axis.text.y = element_text(size = 10),
axis.title = element_text(size = 12),
plot.title = element_text(size = 14, hjust = 0.5)
)
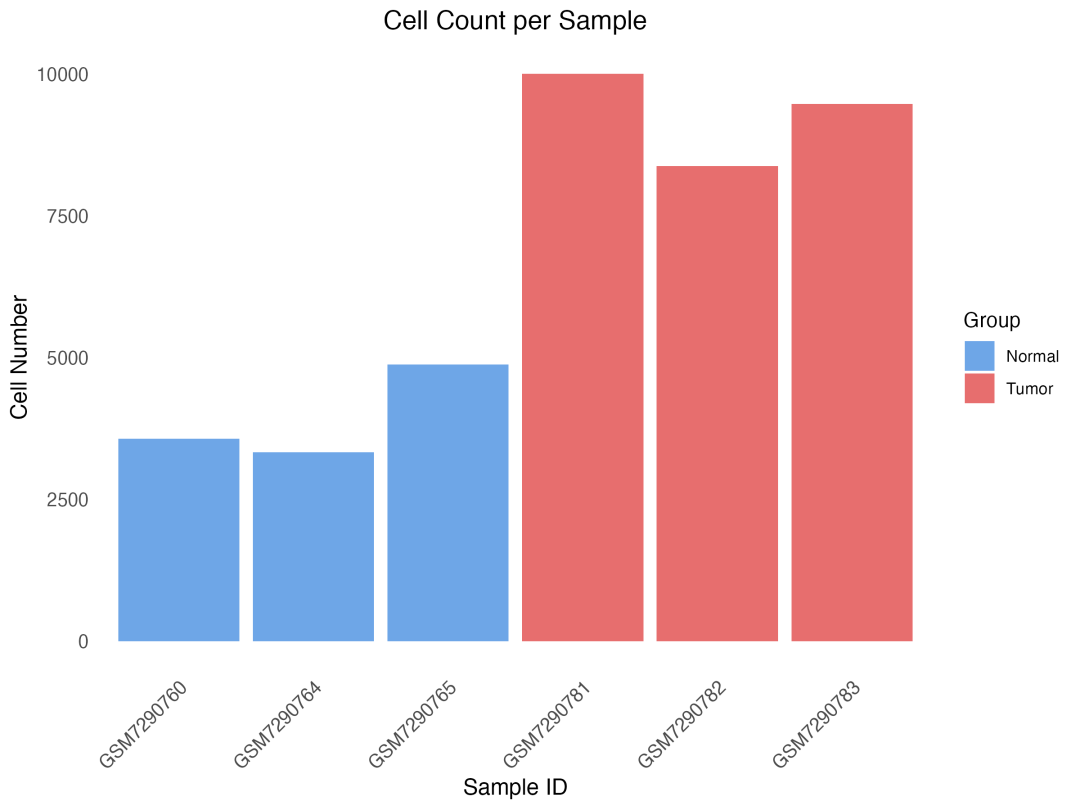
图 1 样本细胞数统计柱状图:展示了每个样本的细胞数量分布,可以看出肿瘤样本的细胞数量明显高于正常样本。
通过可视化分析,我们可以发现几个有趣的现象。首先,肿瘤组的细胞数量(27,863个)明显高于正常组(11,780个),这可能反映了肿瘤组织中更高的细胞密度或更强的细胞增殖活性。其次,不同样本间的细胞数量存在一定差异,这在单细胞实验中是正常现象,可能由组织取样、细胞分离效率、测序深度等多种因素造成。
数据合并后的总体统计显示,我们成功整合了39,643个细胞和38,224个基因,形成了一个相当规模的数据集。这个数据规模足以支持后续的细胞类型识别、差异表达分析和功能富集等多种分析。
在预处理阶段,还需要关注基因注释的一致性。从结果可以看出,正常样本和肿瘤样本的基因数量略有不同(33,538 vs 36,601),这可能是由于不同批次测序或注释版本差异造成的。Seurat的merge函数会自动处理这种情况,只保留所有样本共同的基因,确保后续分析的一致性。
总结
-
文件整理是关键 :正确的10X文件结构是成功读取数据的前提,建议采用样本-文件夹的组织方式 -
逐步读取策略 :面对多样本数据时,逐个读取再合并比直接批量读取更稳妥 -
元数据管理 :及时添加分组信息和样本标识,为后续分析打好基础 -
质量评估 :通过统计图表了解数据的基本特征,识别潜在的批次效应或异常样本
数据读取和预处理看似简单,但是这个步骤很重要。正确的预处理能避免后续分析中的技术问题,如果没处理好,后面可能会白做,容易被审稿人质疑。
完整代码联系老师免费获取(备注“单细胞转录组分析”)

以上就是今天的内容,希望对你有帮助!欢迎点赞、在看、关注、转发。