![[单细胞转录组分析系列教程 2] 怎么做单细胞质控过滤?](/media/blog/covers/cover_20250820.jpg)
大家早上好,今天继续将 单细胞转录组分析。
单细胞测序技术虽然强大,但也容易产生各种技术噪音和低质量数据。在实际分析中,我们经常遇到这样的情况:部分细胞因为细胞膜破损导致大量线粒体RNA泄漏,部分细胞因为裂解不充分导致检测到的基因数量过少,还有一些可能是细胞碎片或双细胞。如果不进行严格的质量控制,这些低质量数据会严重影响后续的聚类分析和生物学解释。本文将详细介绍单细胞数据质控的核心策略和实用技巧。
# 加载必要的包
library(Seurat)
library(ggplot2)
library(dplyr)
library(patchwork)
# 加载数据(基于第一步读取的数据)
data_path <- "../GSE231559_RAW/"
sample_dirs <- list.files(data_path, full.names = TRUE)
sample_dirs <- sample_dirs[dir.exists(sample_dirs)][1] # 取第一个样本演示
counts <- Read10X(data.dir = sample_dirs)
scRNA <- CreateSeuratObject(counts, min.cells = 0, min.features = 0, project = "QC_Demo")
print(paste("数据加载完成,细胞数:", ncol(scRNA), "基因数:", nrow(scRNA)))
质控的第一步是计算关键的质量指标。在单细胞转录组分析中,有几个核心指标可以反映细胞的质量状态:UMI总数(nCount_RNA)反映了细胞的转录活跃程度,检测基因数(nFeature_RNA)体现了捕获效率,线粒体基因百分比则是细胞完整性的重要指标。
线粒体基因百分比尤其重要,因为当细胞膜受损时,胞质RNA会迅速降解,而线粒体RNA因为有双分子膜保护相对稳定,导致线粒体基因比例异常升高。这是识别死细胞或受损细胞的经典标志。
# 检测线粒体基因
mt_genes <- grep("^[M,m][T,t]-", rownames(scRNA), value = TRUE)
print(paste("检测到线粒体基因:", length(mt_genes)))
# 检测核糖体基因
ribo_genes <- grep("^R[P,p][SL,sl]", rownames(scRNA), value = TRUE)
print(paste("检测到核糖体基因:", length(ribo_genes)))
# 计算线粒体基因百分比
scRNA[["percent.mt"]] <- PercentageFeatureSet(scRNA, pattern = "^[M,m][T,t]-")
# 计算核糖体基因百分比
scRNA[["percent.ribo"]] <- PercentageFeatureSet(scRNA, pattern = "^R[P,p][SL,sl]")
计算完质控指标后,我们需要通过可视化来了解数据的分布特征。小提琴图是展示单细胞质控指标的经典方法,它不仅显示了数据的分布范围,还能揭示分布的形状特征。
从我们的分析结果可以看出,该样本包含3,570个细胞,检测到13个线粒体基因和104个核糖体基因。UMI总数的中位数为4,626,这表明大部分细胞具有合理的转录活跃度。线粒体基因百分比的中位数为10.45%,这在正常的生理范围内。
# 图1: 小提琴图展示质控指标分布
p1 <- VlnPlot(scRNA, features = c("nCount_RNA", "nFeature_RNA", "percent.mt", "percent.ribo"),
ncol = 4, pt.size = 0.1) +
theme(axis.text.x = element_text(angle = 0))
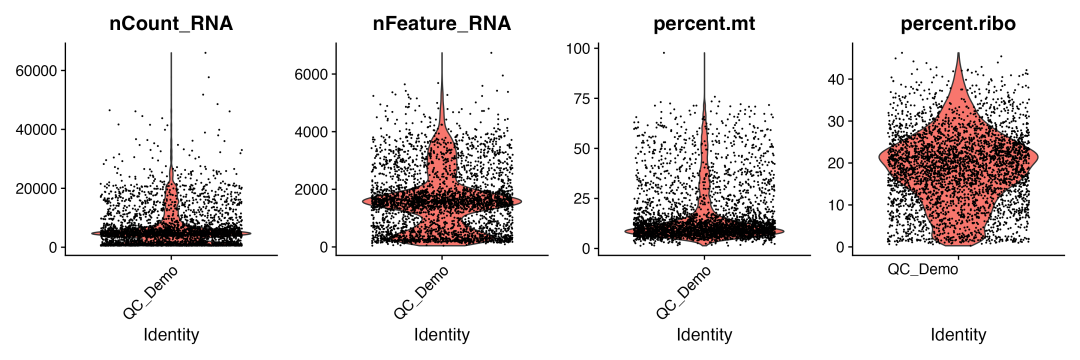
图 1 过滤前质控指标分布:展示了UMI总数、检测基因数、线粒体基因百分比和核糖体基因百分比的分布特征。
从小提琴图可以看出几个重要信息:首先,UMI总数和检测基因数都呈现右偏分布,这是单细胞数据的典型特征。大部分细胞集中在较低的数值区间,少数细胞具有异常高的数值。其次,线粒体基因百分比显示存在一个长尾分布,提示有部分细胞的线粒体基因比例异常高,这些可能是受损或死亡的细胞。
散点图能够进一步揭示不同质控指标之间的相关性,这对于理解细胞状态和设定过滤阈值非常有用。
# 图2: 散点图展示质控指标相关性
plot1 <- FeatureScatter(scRNA, feature1 = "nCount_RNA", feature2 = "nFeature_RNA",
raster = FALSE) +
geom_smooth(method = "lm", se = FALSE, color = "red", size = 0.5) +
theme_minimal() + NoLegend()
plot2 <- FeatureScatter(scRNA, feature1 = "nCount_RNA", feature2 = "percent.mt",
raster = FALSE) +
geom_smooth(method = "lm", se = FALSE, color = "red", size = 0.5) +
theme_minimal() + NoLegend()
plot3 <- FeatureScatter(scRNA, feature1 = "nCount_RNA", feature2 = "percent.ribo",
raster = FALSE) +
geom_smooth(method = "lm", se = FALSE, color = "red", size = 0.5) +
theme_minimal() + NoLegend()
p2 <- plot1 + plot2 + plot3
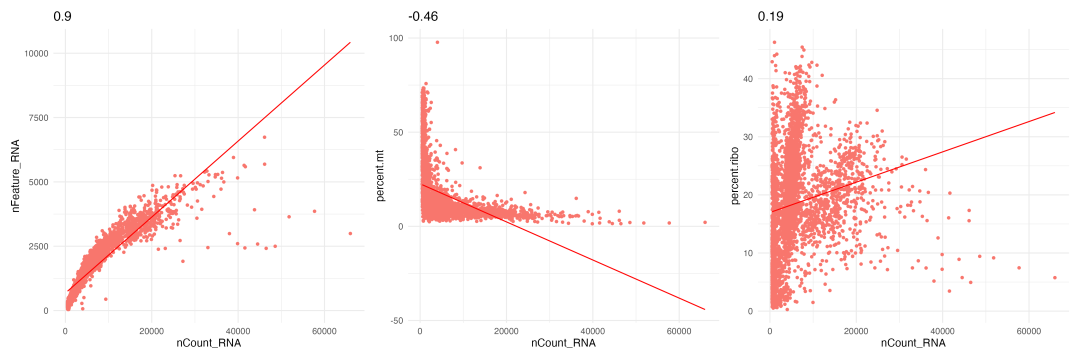
图 2 质控指标相关性散点图:展示了UMI总数与检测基因数、线粒体基因百分比、核糖体基因百分比之间的关系。
散点图分析揭示了几个重要的生物学规律。UMI总数与检测基因数之间存在强正相关,这符合生物学预期——转录更活跃的细胞通常能检测到更多的基因。而UMI总数与线粒体基因百分比之间呈现负相关趋势,这表明转录活跃的健康细胞通常具有较低的线粒体基因比例。
设定过滤阈值是质控过程中最关键的步骤。过于宽松的阈值会保留低质量细胞,影响分析结果;过于严格的阈值则可能误删有用的细胞类型,特别是那些天然具有低转录活跃度的细胞类型。
# 使用MAD方法计算异常值阈值
# 线粒体基因百分比
mt_mad <- mad(scRNA$percent.mt)
mt_upper <- median(scRNA$percent.mt) + 3 * mt_mad
print(paste("线粒体基因上限阈值:", round(mt_upper, 2), "%"))
# 特征基因数
nFeature_mad <- mad(scRNA$nFeature_RNA)
nFeature_lower <- max(200, median(scRNA$nFeature_RNA) - 3 * nFeature_mad)
nFeature_upper <- median(scRNA$nFeature_RNA) + 3 * nFeature_mad
# UMI总数
nCount_mad <- mad(scRNA$nCount_RNA)
nCount_upper <- median(scRNA$nCount_RNA) + 3 * nCount_mad
# 应用过滤
scRNA_filtered <- subset(scRNA,
subset = nFeature_RNA >= nFeature_lower &
nFeature_RNA <= nFeature_upper &
nCount_RNA <= nCount_upper &
percent.mt <= mt_upper)
我们采用了基于中位数绝对偏差(MAD)的方法来设定阈值,这是一种稳健的统计方法,不易受到极端值的影响。具体而言,我们将线粒体基因百分比的上限设为25.25%,特征基因数的范围设为200-4504,UMI总数的上限设为16,151。
这种方法的优势在于它能够自适应数据的分布特征,避免了硬编码阈值可能带来的问题。对于不同的细胞类型、组织来源或实验条件,MAD方法都能提供相对合理的阈值。
# 图3: 过滤后小提琴图
p3 <- VlnPlot(scRNA_filtered, features = c("nCount_RNA", "nFeature_RNA", "percent.mt", "percent.ribo"),
ncol = 4, pt.size = 0.1) +
theme(axis.text.x = element_text(angle = 0))
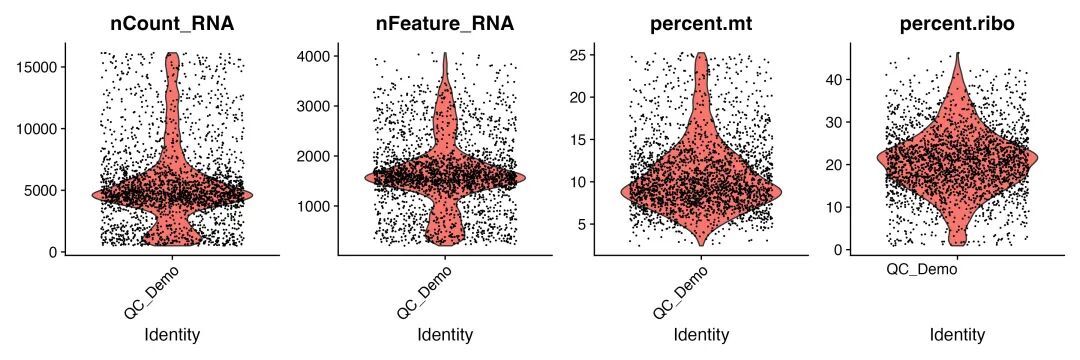
图 3 过滤后质控指标分布:展示了质控过滤后各项指标的分布改善情况。
过滤结果显示,我们从原始的3,570个细胞中保留了2,611个高质量细胞,保留率为73.14%。这个保留率在单细胞分析中是合理的,既保证了数据质量,又尽可能地保留了有用信息。
过滤后的数据质量明显改善:线粒体基因百分比的异常高值被去除,数据分布更加集中;UMI总数和检测基因数的分布也更加正常,极端异常值被有效过滤。
# 图4: 过滤前后对比
comparison_stats <- data.frame(
Stage = c("Before", "After"),
nCells = c(ncol(scRNA), ncol(scRNA_filtered)),
Median_nCount = c(median(scRNA$nCount_RNA), median(scRNA_filtered$nCount_RNA)),
Median_nFeature = c(median(scRNA$nFeature_RNA), median(scRNA_filtered$nFeature_RNA)),
Median_MT = c(median(scRNA$percent.mt), median(scRNA_filtered$percent.mt))
)
p4 <- comparison_stats %>%
ggplot(aes(x = Stage, y = nCells, fill = Stage)) +
geom_col(alpha = 0.8) +
geom_text(aes(label = nCells), vjust = -0.5, size = 5) +
scale_fill_manual(values = c("Before" = "#FF6B6B", "After" = "#4ECDC4")) +
labs(title = "Cell Number Before and After QC Filtering",
x = "Filtering Stage", y = "Cell Number") +
theme_minimal()
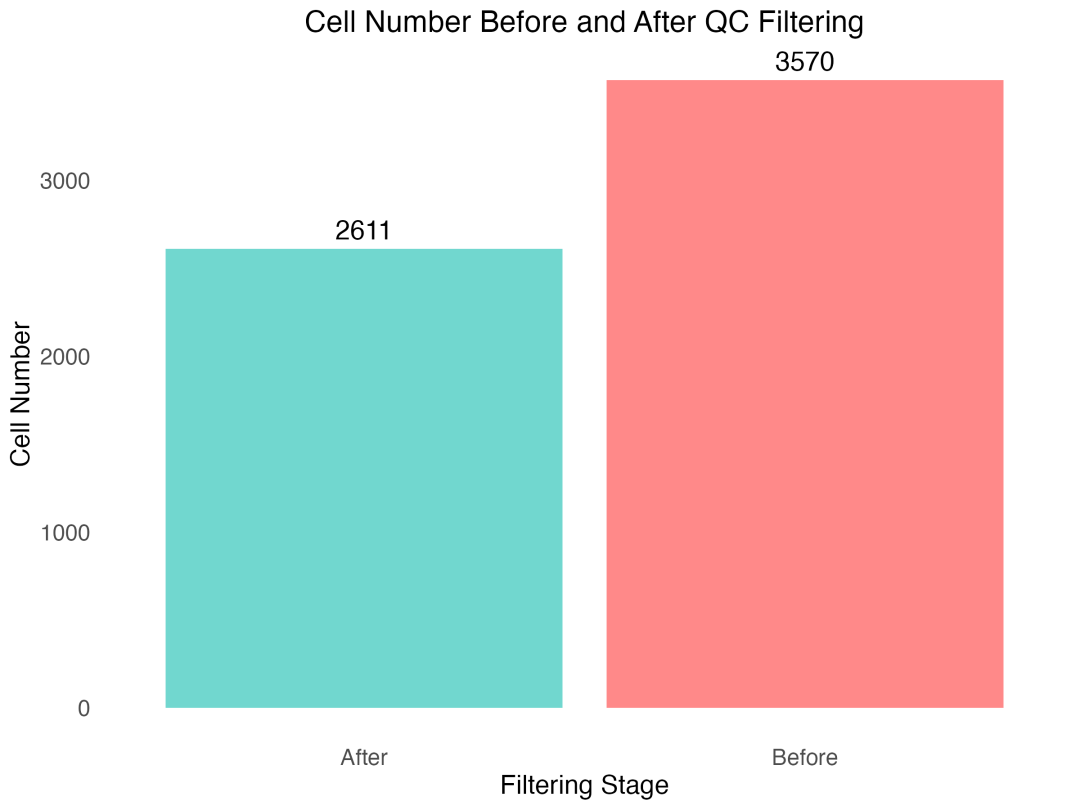
图 4 过滤前后细胞数量对比:直观展示了质控过滤对细胞数量的影响。
质控过滤不仅是为了去除低质量数据,更重要的是为后续分析奠定可靠的基础。高质量的数据能够产生更清晰的细胞聚类结果,更准确的差异表达分析,以及更可信的生物学结论。
在实际应用中,质控阈值的设定需要结合具体的生物学背景。例如,某些细胞类型(如红细胞前体细胞)天然具有较高的线粒体基因表达;某些组织(如肌肉组织)的细胞可能具有更高的基础转录水平。因此,建议在设定阈值时参考相关文献,并结合实验的具体情况进行调整。
总结
-
指标计算是基础 :准确计算线粒体基因、核糖体基因百分比等关键质控指标 -
可视化是关键 :通过小提琴图和散点图全面了解数据分布和相关性特征 -
阈值设定需谨慎 :推荐使用MAD等稳健统计方法,避免过度依赖硬编码阈值 -
结果评估很重要 :关注过滤前后的数据质量变化和保留率,确保平衡质量与数量
质控过滤是单细胞分析中最需要经验和判断力的步骤之一。良好的质控策略不仅能提升数据质量,更能为后续的生物学发现提供坚实基础。记住,质控的目标不是获得最多的细胞,而是获得最可靠的细胞。