![[单细胞转录组分析系列教程 3] 怎么做单细胞标准化和变异基因识别?](/media/blog/covers/cover_20250821.jpg)
大家早上好,今天继续讲解单细胞转录组分析系列。
在单细胞转录组数据分析中,原始的UMI计数数据存在明显的技术偏差和生物学噪音。不同细胞的测序深度差异很大,基因表达水平的分布也极不均匀,这些问题如果不加处理就直接进行下游分析,会严重影响聚类结果的准确性和生物学解释的可靠性。数据标准化和变异基因识别是解决这些问题的关键步骤,也是连接质控过滤和降维分析的重要桥梁。
# 加载必要的包
library(Seurat)
library(ggplot2)
library(dplyr)
# 加载质控后的数据(基于前面步骤的数据)
# 合并layers以兼容Seurat v5
scRNA <- JoinLayers(scRNA)
# 简单质控过滤
scRNA[["percent.mt"]] <- PercentageFeatureSet(scRNA, pattern = "^[M,m][T,t]-")
scRNA <- subset(scRNA, subset = nFeature_RNA > 200 & nFeature_RNA < 5000 & percent.mt < 20)
print(paste("质控后数据:", ncol(scRNA), "个细胞,", nrow(scRNA), "个基因"))
单细胞数据标准化的核心目标是消除技术性的细胞间差异,同时保留生物学上有意义的变异。原始的UMI计数数据具有强烈的技术偏差——不同细胞的总UMI数可能相差数十倍,这种差异主要来源于细胞捕获效率、反转录效率和测序深度的不同,而非真实的生物学差异。
从我们的分析结果可以看出,质控后的数据包含9,519个细胞和33,538个基因。数据具有典型的稀疏性特征,94.02%的位置为零值,这反映了单细胞数据的天然特点——大部分基因在特定细胞中不表达或表达量极低。平均每个细胞检测到2,005个基因,平均UMI计数为7,229,这些指标表明数据质量良好。
# 检查标准化前的数据特征
raw_counts <- GetAssayData(scRNA, layer = "counts")
total_umis <- Matrix::colSums(raw_counts)
detected_genes <- Matrix::colSums(raw_counts > 0)
print("标准化前数据统计:")
print(paste("平均UMI计数:", round(mean(total_umis), 2)))
print(paste("平均检测基因数:", round(mean(detected_genes), 2)))
print(paste("数据稀疏性:", round(100 * (1 - sum(raw_counts > 0) / length(raw_counts)), 2), "%"))
# 执行LogNormalize标准化
scRNA <- NormalizeData(scRNA, normalization.method = "LogNormalize", scale.factor = 10000)
print("数据标准化完成:LogNormalize方法,缩放因子10000")
LogNormalize是目前最广泛使用的标准化方法。它的工作原理是:首先计算每个细胞的总UMI数,然后将每个基因的表达量除以该细胞的总UMI数,接着乘以缩放因子(通常是10,000),最后进行自然对数转换(加1以避免对数零值)。这种方法能有效地将不同细胞的表达量调整到可比较的水平。
标准化过程中的对数转换特别重要。它不仅压缩了数据的动态范围,还使得数据分布更接近正态分布,这对于基于欧几里得距离的聚类算法和主成分分析都是有益的。缩放因子10,000的选择是为了保持数值在合理范围内,便于后续计算和解释。
数据标准化完成后,下一个关键步骤是识别高变异基因。在单细胞数据中,并非所有基因都包含有用的生物学信息。大量基因在所有细胞中的表达都比较稳定,这些基因虽然可能在维持细胞基本功能方面很重要,但在区分不同细胞类型方面作用有限。
# 识别高变异基因
scRNA <- FindVariableFeatures(scRNA, selection.method = "vst", nfeatures = 2000)
# 获取变异基因信息
variable_features <- VariableFeatures(scRNA)
print(paste("成功识别", length(variable_features), "个高变异基因"))
# 获取前10个最变异的基因
top10 <- head(VariableFeatures(scRNA), 10)
print("前10个最变异基因:")
print(top10)
# 生成变异基因识别图
plot1 <- VariableFeaturePlot(scRNA)
plot2 <- LabelPoints(plot = plot1, points = top10, repel = TRUE)
library(patchwork)
p_combined <- plot1 + plot2
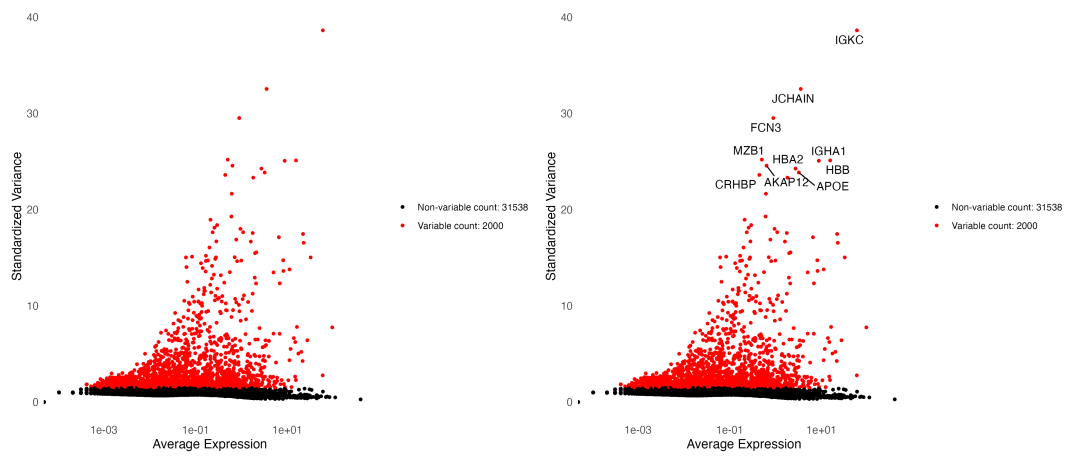
图 变异基因识别结果:左图显示所有基因的平均表达量与标准化方差的关系,红点标记高变异基因;右图额外标注了变异程度最高的10个基因。
我们成功识别了2,000个高变异基因,这个数量在单细胞分析中是标准的选择。有趣的是,前10个最变异的基因包括IGKC、JCHAIN等免疫球蛋白相关基因,以及HBB、HBA2等血红蛋白基因,这反映了肝脏组织中复杂的细胞组成,包括B细胞、浆细胞和可能的红细胞前体细胞。
VST(variance stabilizing transformation)方法是目前推荐的变异基因识别方法。它通过拟合平均表达量与方差之间的关系,识别那些方差显著高于预期的基因。这种方法相比简单的方差排序更加稳健,能够避免高表达基因自动被选为变异基因的偏差,确保在不同表达水平上都能识别到真正的变异基因。
# 变异基因统计分析
hvf_info <- HVFInfo(scRNA)
hvf_stats <- hvf_info[variable_features, ]
print("=== 变异基因特征分析 ===")
print("平均表达水平统计:")
print(summary(hvf_stats$mean))
print("标准化方差统计:")
print(summary(hvf_stats$variance.standardized))
# 分析变异基因的组成
mt_variable <- sum(grepl("^[M,m][T,t]-", variable_features))
ribo_variable <- sum(grepl("^R[P,p][SL,sl]", variable_features))
print("=== 变异基因组成分析 ===")
print(paste("线粒体基因数量:", mt_variable, "个"))
print(paste("核糖体基因数量:", ribo_variable, "个"))
print(paste("其他基因数量:", length(variable_features) - mt_variable - ribo_variable, "个"))
# 检查重要的基因类型
immune_genes <- sum(grepl("^IG[KLH]|^CD[0-9]|^HLA-", variable_features))
print(paste("免疫相关基因数量:", immune_genes, "个"))
统计分析显示,变异基因的平均表达水平范围很广,从极低表达(0.00042)到高表达(95.09)都有涵盖,这表明VST方法成功地在不同表达水平上识别了变异基因。标准化方差的范围为1.48-38.63,中位数为2.09,这些数值确认了所选基因确实具有显著的细胞间变异。
令人欣慰的是,我们的变异基因中线粒体基因数量为0,核糖体基因仅有1个(占比0.05%),这表明数据质量良好,变异基因主要由真正的生物学相关基因组成。有112个基因被识别为免疫相关基因,这与肝脏作为重要免疫器官的生物学特性一致。另外还识别出12个应激反应基因,这可能反映了细胞在组织处理过程中的应激状态。
变异基因的选择是后续所有分析的基础。在主成分分析、聚类分析等步骤中,通常只使用这些高变异基因,这不仅能显著提高计算效率,更重要的是能提高分析结果的生物学相关性。通过专注于那些真正驱动细胞异质性的基因,我们能够更准确地识别不同的细胞类型和细胞状态,避免被技术噪音和管家基因的稳定表达所干扰。
正确的标准化和变异基因识别为后续的降维分析、聚类识别和差异表达分析奠定了坚实基础。LogNormalize标准化消除了技术性的测序深度差异,使得不同细胞间的表达水平具有可比性;VST方法识别的高变异基因则确保我们专注于最有信息价值的基因子集,为发现生物学意义提供了最佳起点。
总结
-
标准化是基础 :LogNormalize方法能有效消除技术偏差,对数转换改善数据分布特征 -
变异基因选择很关键 :VST方法能在各表达水平识别变异基因,避免高表达偏差 -
质量评估不可缺 :检查变异基因组成,确保生物学相关基因占主导地位 -
参数设置需合理 :2000个变异基因通常足够捕获主要的生物学变异,兼顾信息保留和计算效率
数据标准化和变异基因识别虽然看似技术性较强,但其本质是在为后续的生物学发现做准备。正确的标准化能让我们专注于真正的生物学差异,而精确的变异基因识别则确保我们不会在噪音中迷失方向,为揭示细胞异质性的生物学机制打下坚实基础。