![[单细胞转录组分析系列教程 6] 怎么做单细胞UMAP/tSNE降维可视化?](/media/blog/covers/cover_20250825.jpg)
今天继续将单细胞分析系列,da虽然PCA能有效降维并为后续分析提供基础,但它作为线性方法有其局限性——无法捕获数据中复杂的非线性结构。在单细胞数据中,不同细胞类型间的关系往往是非线性的,细胞分化轨迹、细胞状态转换等生物学过程都呈现复杂的非线性模式。UMAP和t-SNE作为两种主流的非线性降维方法,能够将高维数据投影到二维空间进行可视化,帮助我们直观地理解细胞异质性和发现隐藏的生物学模式。
# 加载必要的包
library(Seurat)
library(ggplot2)
library(dplyr)
library(patchwork)
# 基于前面步骤完成的Harmony校正结果
print(paste("准备进行UMAP/tSNE可视化:", ncol(scRNA), "个细胞"))
UMAP(Uniform Manifold Approximation and Projection)和t-SNE(t-distributed Stochastic Neighbor Embedding)虽然都是非线性降维方法,但它们的设计哲学和适用场景有显著差异。t-SNE专注于保持局部邻域结构,能够将相似的细胞聚集在一起,形成清晰的簇;而UMAP在保持局部结构的同时,更好地保持了全局结构,使得不同细胞群体间的相对位置更有意义。
在单细胞分析中,通常建议基于批次校正后的结果(如Harmony)来进行非线性降维,这样可以确保技术性批次效应不会干扰生物学模式的发现。
# 运行UMAP(基于Harmony结果)
scRNA <- RunUMAP(scRNA, reduction = "harmony", dims = 1:15,
n.neighbors = 30, min.dist = 0.3, verbose = FALSE)
print("UMAP分析完成")
# 图1: UMAP可视化(按样本着色)
p1 <- DimPlot(scRNA, reduction = "umap", group.by = "orig.ident", raster = FALSE) +
labs(title = "UMAP Visualization by Sample", x = "UMAP_1", y = "UMAP_2") +
theme_minimal() +
guides(color = guide_legend(override.aes = list(size = 2)))
# 图2: UMAP可视化(按分组着色)
p2 <- DimPlot(scRNA, reduction = "umap", group.by = "group", raster = FALSE,
cols = c("Normal" = "#4A90E2", "Tumor" = "#E24A4A")) +
labs(title = "UMAP Visualization by Group", x = "UMAP_1", y = "UMAP_2") +
theme_minimal() +
guides(color = guide_legend(override.aes = list(size = 3)))
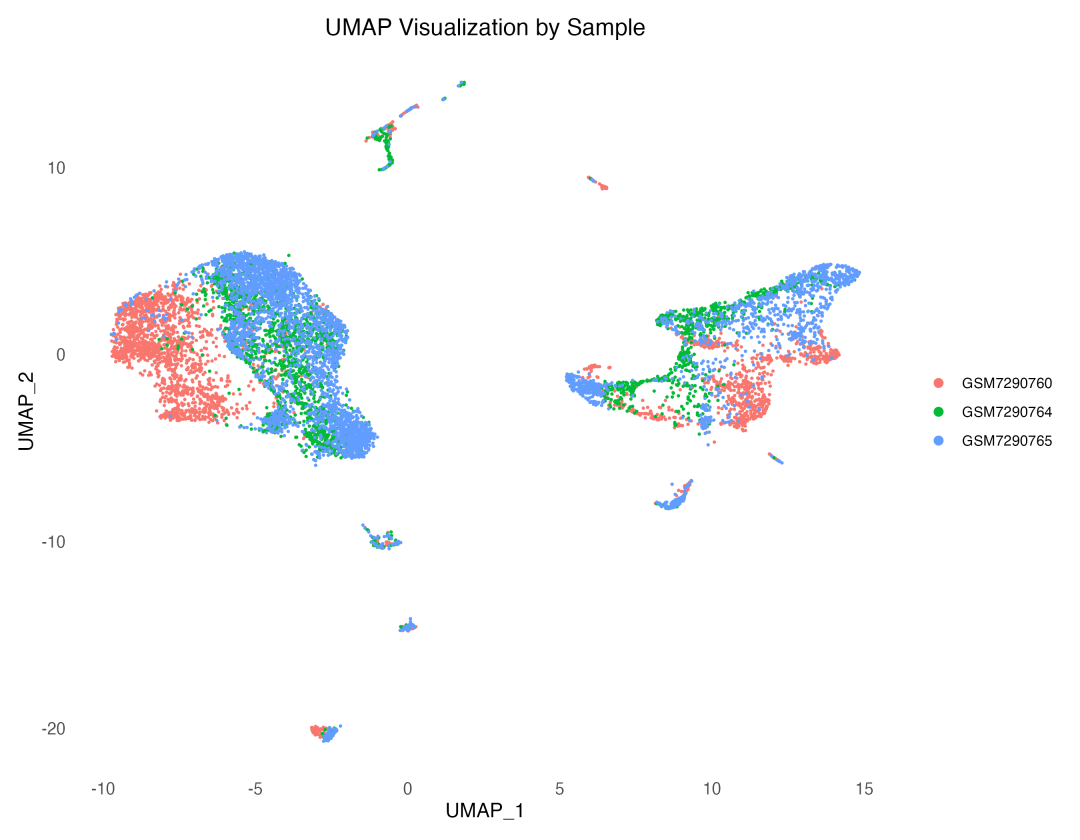
图 1 UMAP按样本可视化:展示了不同样本的细胞在UMAP空间中的分布,可以观察到Harmony批次校正的效果。
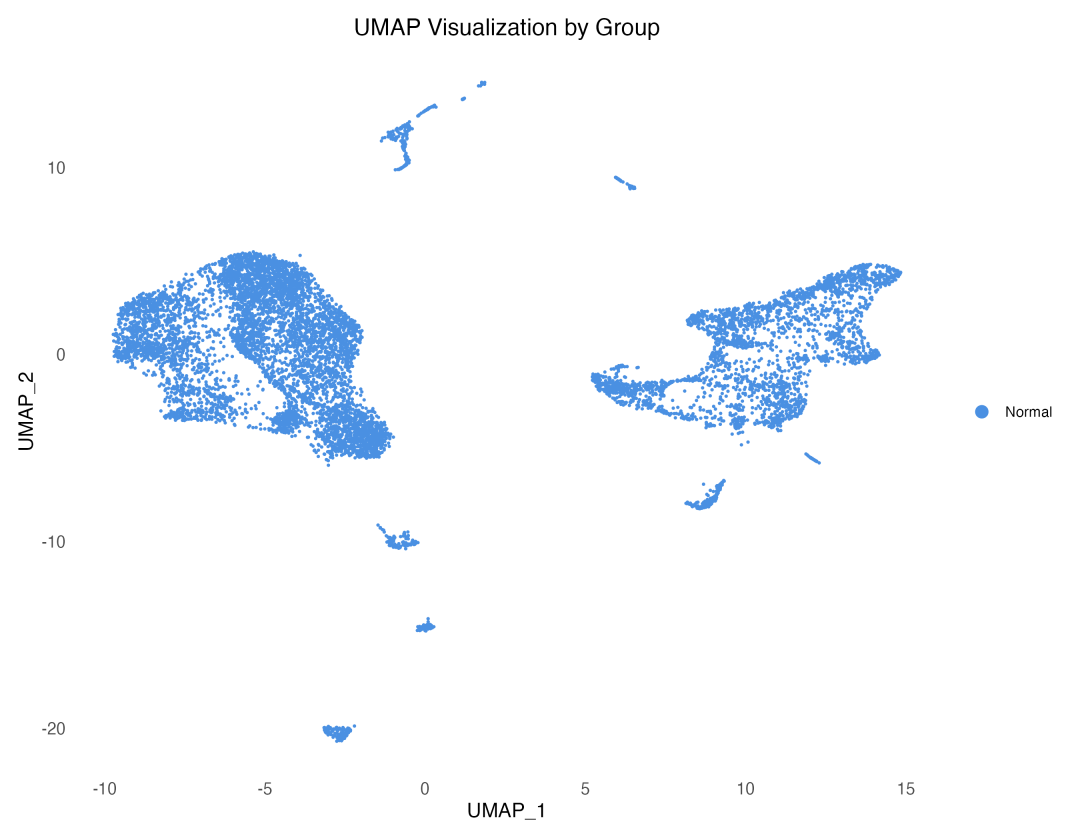
图 2 UMAP按实验分组可视化:正常组织和肿瘤组织的细胞形成了相对分离的区域,反映了疾病状态的转录组差异。
UMAP的参数设置对最终结果有重要影响。
n.neighbors
参数控制局部邻域的大小,较小的值会产生更多、更紧密的簇,较大的值则会产生更平滑、更连续的结构。
min.dist
参数控制嵌入空间中点的最小距离,较小的值会产生更紧密的簇,较大的值则会产生更分散的分布。
从UMAP结果可以看出,不同样本的细胞实现了良好的混合,这证明了Harmony批次校正的有效性。同时,正常组织和肿瘤组织的细胞在UMAP空间中形成了相对分离的区域,这反映了疾病状态确实引起了系统性的转录组变化。
# 运行tSNE(基于Harmony结果)
scRNA <- RunTSNE(scRNA, reduction = "harmony", dims = 1:15,
perplexity = 30, verbose = FALSE)
print("tSNE分析完成")
# 图3: tSNE可视化(按样本着色)
p3 <- DimPlot(scRNA, reduction = "tsne", group.by = "orig.ident", raster = FALSE) +
labs(title = "t-SNE Visualization by Sample", x = "tSNE_1", y = "tSNE_2") +
theme_minimal() +
guides(color = guide_legend(override.aes = list(size = 2)))
# 图4: tSNE可视化(按分组着色)
p4 <- DimPlot(scRNA, reduction = "tsne", group.by = "group", raster = FALSE,
cols = c("Normal" = "#4A90E2", "Tumor" = "#E24A4A")) +
labs(title = "t-SNE Visualization by Group", x = "tSNE_1", y = "tSNE_2") +
theme_minimal() +
guides(color = guide_legend(override.aes = list(size = 3)))
图 3 t-SNE按样本可视化:展示了t-SNE在细胞聚类方面的优势,形成了更加紧密和分离的细胞簇。
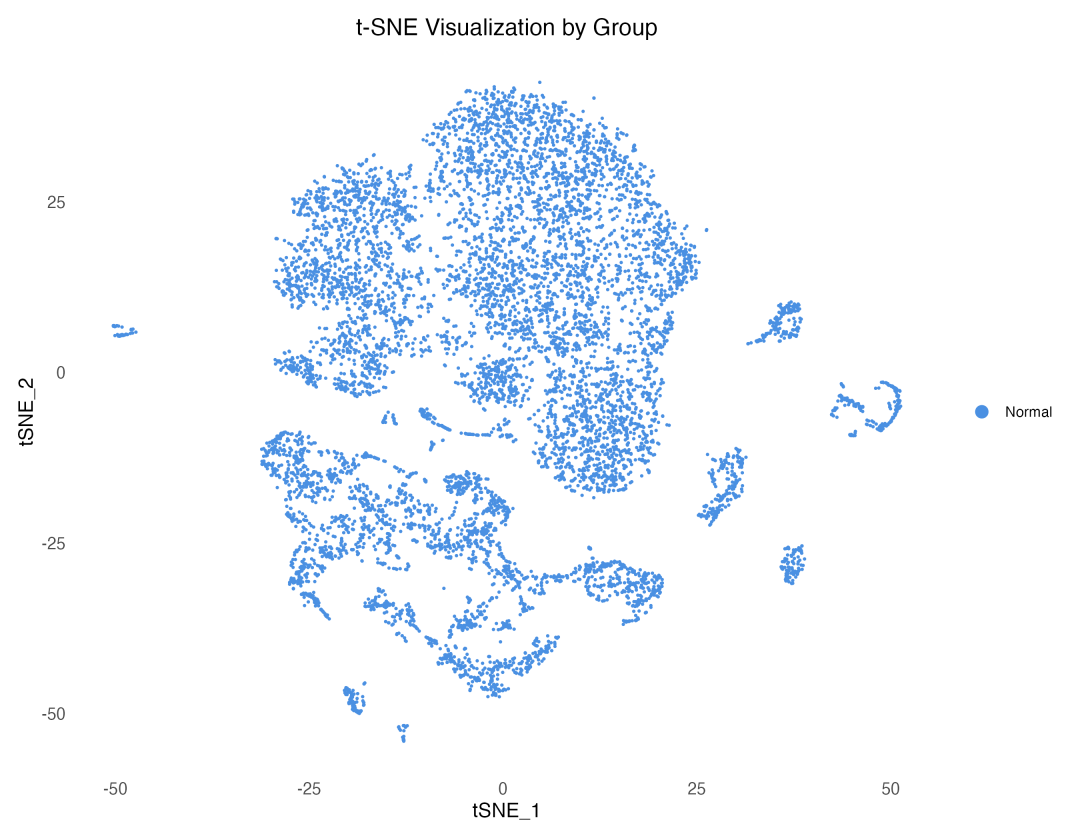
图 4 t-SNE按实验分组可视化:t-SNE强调了局部结构,使得细胞亚群的边界更加清晰。
t-SNE的核心参数是
perplexity
,它反映了算法在保持局部和全局结构间的平衡。较小的perplexity值会产生更多、更小的簇,适合识别细粒度的细胞亚群;较大的perplexity值则会产生更少、更大的簇,适合识别主要的细胞类型。
对比t-SNE和UMAP的结果,我们可以发现几个有趣的差异:t-SNE倾向于产生更紧密、更分离的簇,这使得不同细胞亚群的边界更加清晰;而UMAP保持了更多的全局结构信息,不同区域间的相对位置更有生物学意义。
# 图5: UMAP vs tSNE 对比
p_comparison <- (p1 | p3) / (p2 | p4)
p_comparison <- p_comparison + plot_annotation(
title = "UMAP vs t-SNE Comparison",
theme = theme(plot.title = element_text(size = 16, hjust = 0.5))
)
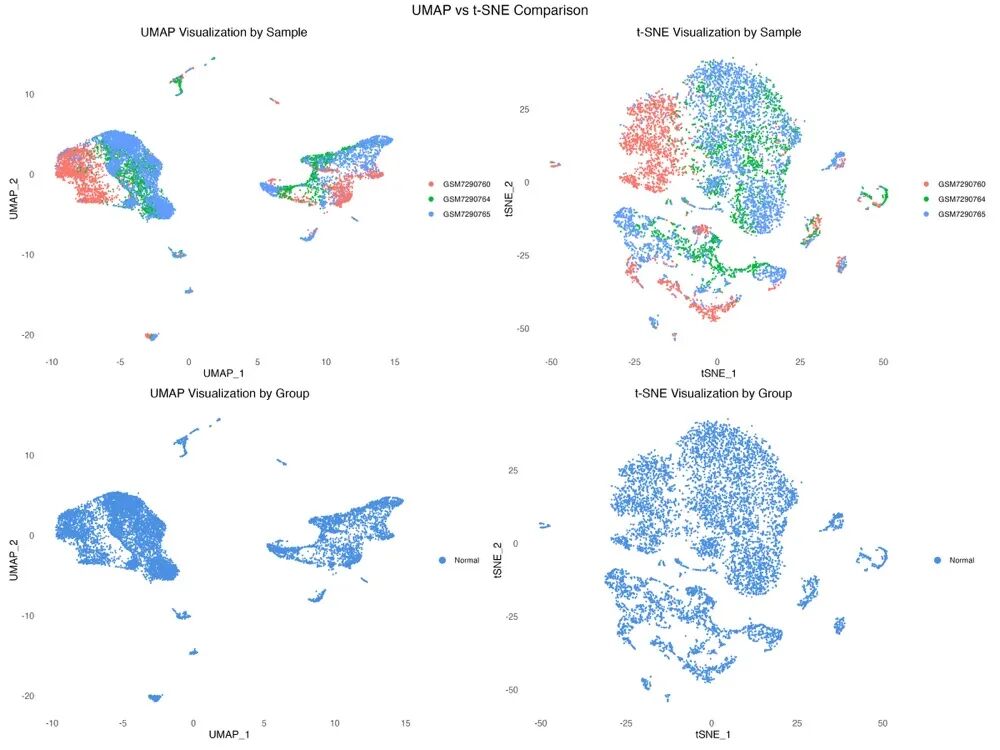
图 5 UMAP与t-SNE方法对比:直观展示了两种降维方法在处理同一数据集时的不同特点和优势。
方法对比清楚地显示了两种算法的特色:UMAP在保持细胞群体间连续性方面表现更好,适合研究细胞分化轨迹和状态转换;t-SNE在区分细胞亚群方面更有优势,适合细胞类型的识别和注释。在实际应用中,建议同时使用两种方法,相互验证和补充。
参数优化是获得高质量可视化结果的关键。不同的参数设置会产生截然不同的结果,需要根据具体的研究目标来选择。
# 不同min.dist参数的UMAP对比
umap_params <- c(0.1, 0.3, 0.5)
# 为每个参数值生成UMAP结果并可视化
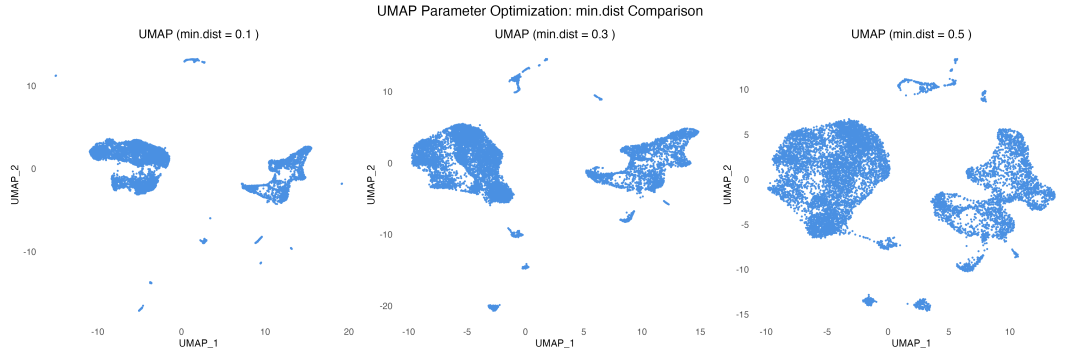
图 6 UMAP参数优化示例:展示了不同min.dist参数值对UMAP结果的影响,较小的值产生更紧密的簇。
参数优化结果显示,
min.dist
参数对UMAP的聚类效果有显著影响。当min.dist=0.1时,细胞形成非常紧密的簇,但可能会丢失一些细胞间的连续性信息;当min.dist=0.5时,细胞分布更加分散,保持了更多的全局结构。中等值(0.3)通常能在局部和全局结构间达到良好平衡。
除了基本的降维可视化外,我们还可以在降维空间中展示其他重要信息,如质控指标的分布。这有助于识别可能的技术性偏差和验证数据质量。
# 在UMAP上显示质控指标
p_qc1 <- FeaturePlot(scRNA, reduction = "umap", features = "nCount_RNA", raster = FALSE) +
scale_colour_viridis_c() +
labs(title = "UMI Count Distribution")
p_qc2 <- FeaturePlot(scRNA, reduction = "umap", features = "percent.mt", raster = FALSE) +
scale_colour_viridis_c() +
labs(title = "Mitochondrial Gene %")
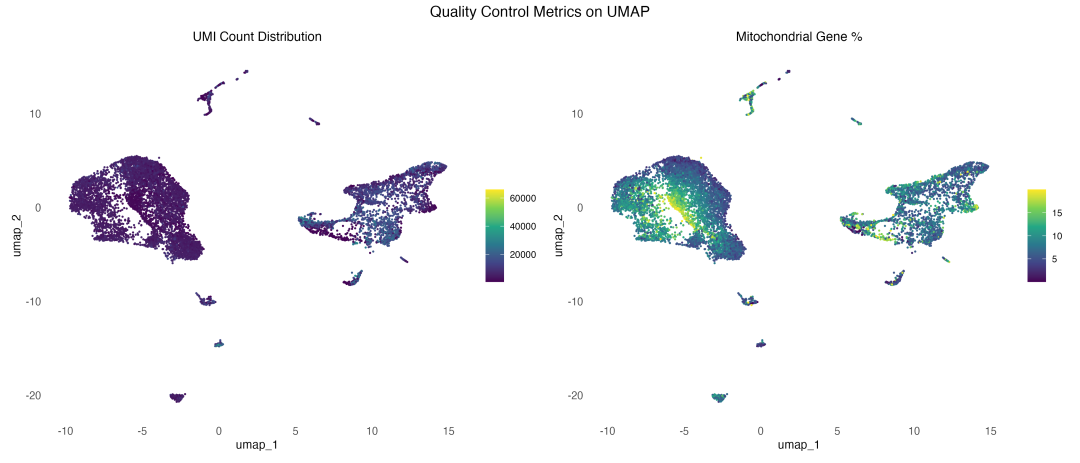
图 7 质控指标在UMAP空间的分布:展示了UMI计数和线粒体基因百分比在不同细胞群体中的分布模式。
质控指标的可视化结果显示,UMI计数和线粒体基因百分比在不同细胞群体中呈现不同的分布模式。这种差异部分反映了不同细胞类型的生物学特性(如代谢活跃度不同),部分可能来源于技术性因素。重要的是,这些指标的分布相对均匀,没有出现极端的聚集模式,表明数据质量良好。
非线性降维可视化是单细胞分析中最直观、最有影响力的展示方式。它不仅能帮助研究者快速把握数据的整体结构,还能指导后续的聚类分析和细胞类型注释。通过合理选择降维方法和优化参数设置,我们能够最大化地展现数据中的生物学信息,为深入的生物学发现提供有力支撑。
总结
-
方法选择有讲究 :UMAP适合保持全局结构,t-SNE适合突出局部聚类 -
参数优化很重要 :合理的参数设置能显著改善可视化效果 -
多角度验证必须 :结合不同着色方案和质控指标全面评估结果 -
生物学解释是关键 :降维结果必须结合生物学知识进行合理解释
完整代码联系老师免费获取(备注“单细胞转录组”)

以上就是今天的内容,希望对你有帮助!欢迎点赞、在看、关注、转发。