![[单细胞转录组分析系列教程 7] 怎么做单细胞聚类分析?](/media/blog/covers/cover_20250827.jpg)
大家早上好,今天继续讲 单细胞转录组分析系列
在单细胞转录组分析中,聚类分析是识别细胞类型和细胞状态的核心步骤。经过降维可视化,我们能够直观地观察到细胞在二维空间中的分布模式,但真正的细胞类型识别还需要依靠无监督聚类算法来实现。聚类分析的目标是将转录组特征相似的细胞归为一组,每个聚类理论上对应一个细胞类型或细胞状态。然而,如何选择合适的聚类参数、如何评估聚类质量、如何确定最优的聚类数量,这些都是需要仔细考虑的关键问题。
# 加载必要的包
library(Seurat)
library(clustree)
library(ggplot2)
library(dplyr)
library(tidyr)
library(patchwork)
# 基于前面步骤完成的Harmony校正结果
print(paste("准备进行聚类分析:", ncol(scRNA), "个细胞"))
现代单细胞聚类分析主要采用基于图的方法,其核心思想是先构建细胞间的邻居图(KNN图),然后在这个图上应用社区发现算法来识别细胞群体。这种方法相比传统的K-means等算法有显著优势:它能够处理任意形状的聚类,对数据的分布假设较少,并且在高维数据中表现稳定。
Seurat中使用的是基于共享最近邻(SNN)的图构建方法,它不仅考虑细胞间的距离,还考虑邻居的重叠程度,这使得聚类结果更加稳定和生物学意义明确。
# 基于Harmony结果构建KNN图
scRNA <- FindNeighbors(scRNA, reduction = "harmony", dims = 1:15, verbose = FALSE)
print("邻居图构建完成")
# 设定不同的分辨率参数进行聚类
resolutions <- c(0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.8, 1.0, 1.2)
# 执行多分辨率聚类
scRNA <- FindClusters(scRNA, resolution = resolutions, verbose = FALSE)
# 查看不同分辨率下的聚类数量
cluster_counts <- sapply(resolutions, function(res) {
length(unique(scRNA@meta.data[, paste0("RNA_snn_res.", res)]))
})
邻居图的构建基于Harmony校正后的前15个成分,这确保了聚类分析不会受到批次效应的干扰。分辨率参数是聚类分析中最关键的参数,它控制聚类的粗细程度。较低的分辨率会产生较少、较大的聚类,适合识别主要的细胞类型;较高的分辨率会产生较多、较小的聚类,适合发现细胞亚型和过渡状态。
从我们的分析结果可以看出,随着分辨率从0.1增加到1.2,聚类数量从11个逐渐增加到23个。这种趋势是正常的,但选择哪个分辨率作为最终结果需要结合生物学知识和数据特征来判断。
Clustree是评估不同分辨率聚类结果的强大工具。它通过构建聚类间的层次关系图,帮助我们理解不同分辨率下聚类的稳定性和分化模式。
# 生成clustree图
p_clustree <- clustree(scRNA, prefix = "RNA_snn_res.")
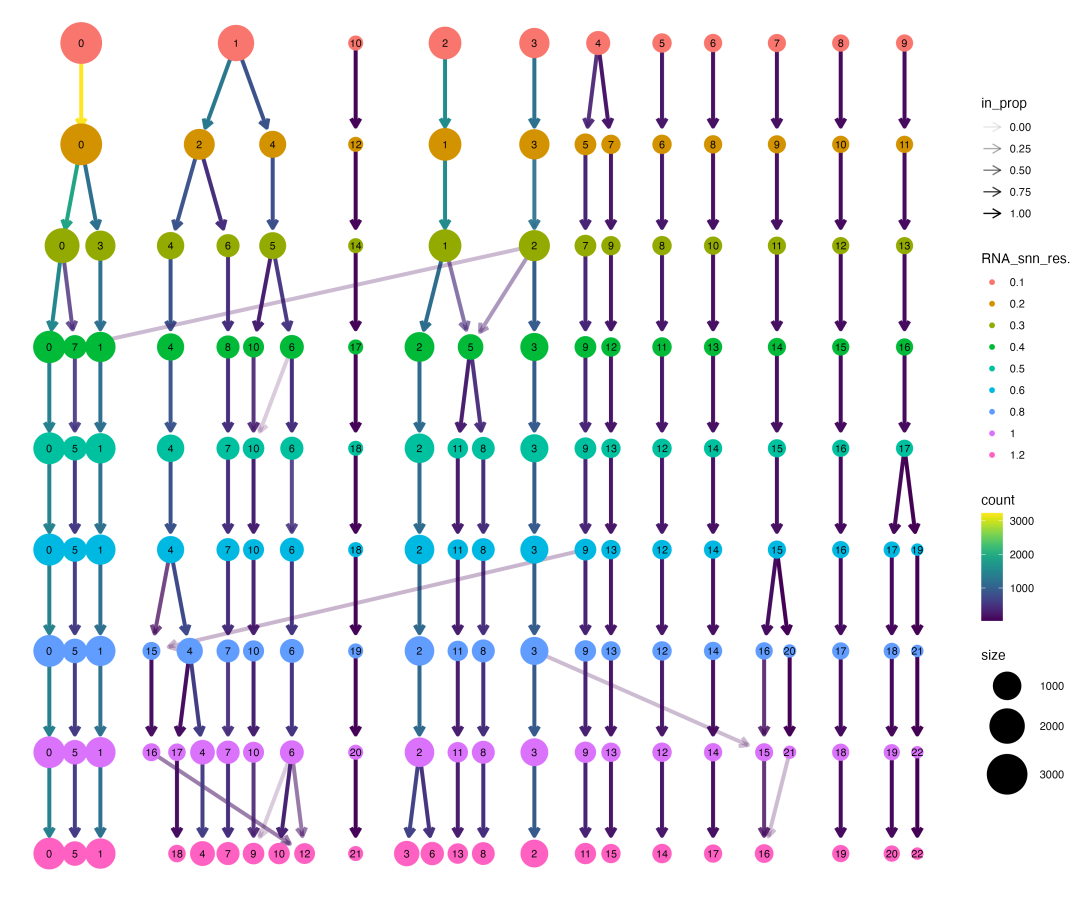
图 1 Clustree聚类关系图:展示了不同分辨率下聚类的演化关系,节点大小表示聚类的细胞数量,连线表示聚类间的关系。
Clustree图提供了选择最优分辨率的重要线索。理想的分辨率应该满足几个条件:聚类稳定(不会在相邻分辨率间频繁分裂合并)、生物学合理(聚类数量符合预期的细胞类型数量)、平衡细致度和简洁性。从图中可以观察到,某些聚类在低分辨率下就已经稳定存在,而另一些则在高分辨率下才出现分化。
基于clustree分析和生物学合理性,我们选择分辨率0.5作为最优参数,它产生了19个聚类,这个数量对于肝脏组织的细胞异质性是合理的。
# 选择最优分辨率0.5
optimal_resolution <- 0.5
Idents(scRNA) <- paste0("RNA_snn_res.", optimal_resolution)
# UMAP上显示聚类结果
p_umap_clusters <- DimPlot(scRNA, reduction = "umap", label = TRUE, label.size = 4, raster = FALSE) +
labs(title = paste("Clustering Results (Resolution =", optimal_resolution, ")"),
x = "UMAP_1", y = "UMAP_2") +
theme_minimal()
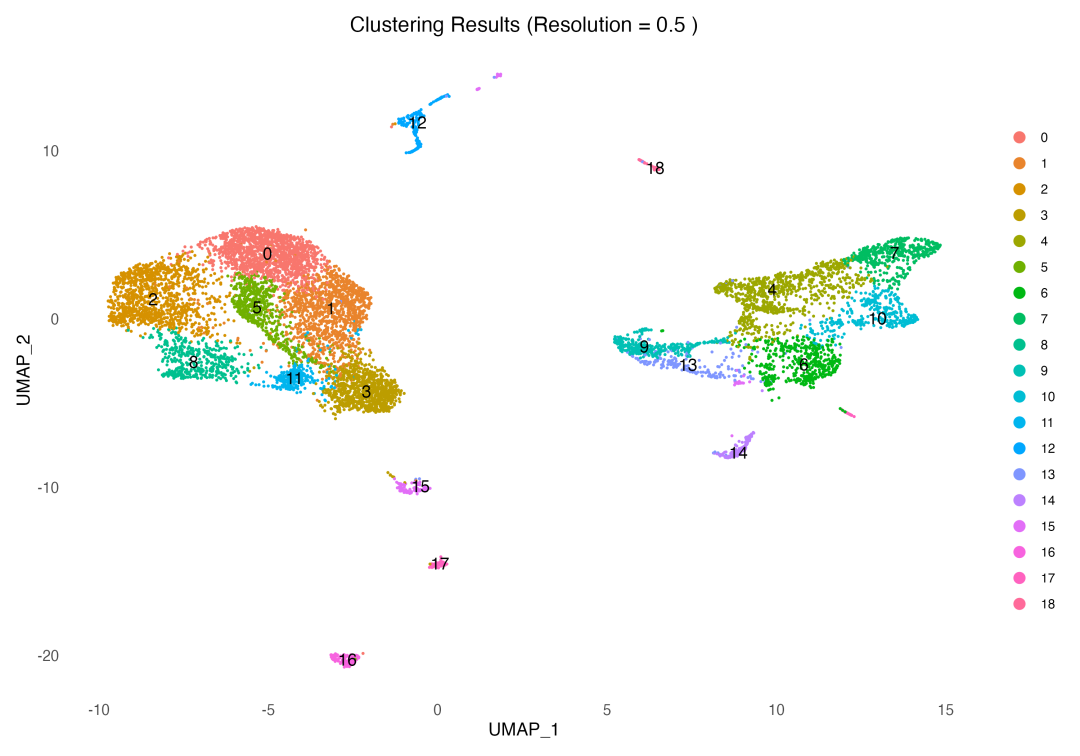
图 2 聚类结果在UMAP上的展示:19个聚类在UMAP空间中形成了清晰的分离,每个聚类都有明确的边界和合理的大小。
聚类结果显示了良好的分离度和连续性。大部分聚类在UMAP空间中形成了紧密的群体,边界清晰,这表明聚类算法成功地识别了转录组特征相似的细胞群体。同时,某些聚类间存在一定的连续性,这可能反映了细胞分化的轨迹或细胞状态的过渡。
为了验证聚类结果的生物学合理性,我们需要检查不同聚类在原始样本和实验分组中的分布情况。
# 按样本和分组展示聚类分布
p_sample <- DimPlot(scRNA, reduction = "umap", group.by = "orig.ident", raster = FALSE) +
labs(title = "Clustering: Sample Distribution")
p_group <- DimPlot(scRNA, reduction = "umap", group.by = "group", raster = FALSE,
cols = c("Normal" = "#4A90E2", "Tumor" = "#E24A4A")) +
labs(title = "Clustering: Group Distribution")
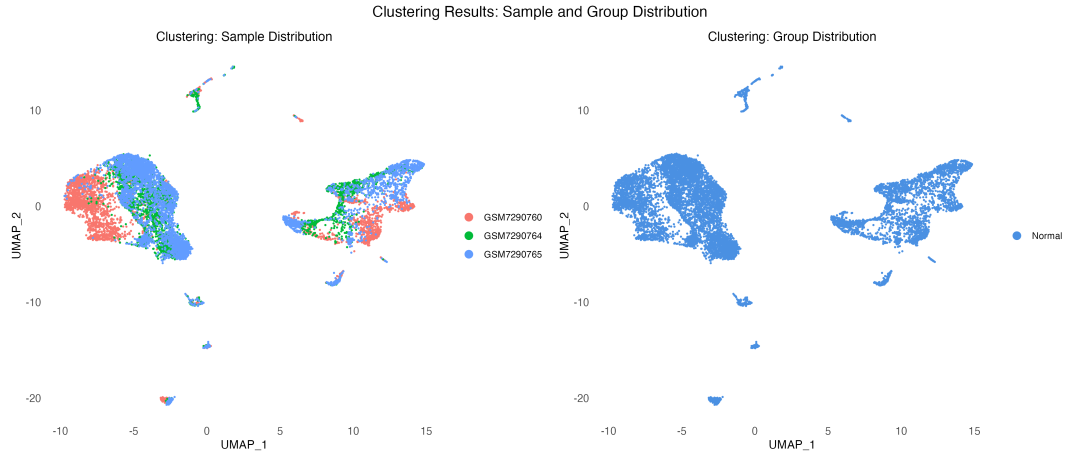
图 3 聚类结果的样本和分组分布验证:左图显示不同样本的细胞在各聚类中的分布,右图显示正常和肿瘤组织的分布模式。
样本分布分析显示,不同样本的细胞在各个聚类中都有分布,这证明了批次校正的有效性和聚类结果的稳定性。如果某些聚类只包含特定样本的细胞,那可能是批次效应而非真正的生物学差异。
分组分布分析揭示了有趣的生物学模式:某些聚类主要包含正常组织的细胞,某些主要包含肿瘤组织的细胞,还有一些聚类包含两种组织的细胞。这种模式是合理的,因为正常和肿瘤组织既有共同的基础细胞类型,也有各自特有的细胞状态。
聚类稳定性是评估聚类质量的重要指标。我们通过比较不同分辨率下聚类大小的分布来评估稳定性。
# 计算不同分辨率下聚类的细胞数分布
# 对比分辨率0.3、0.5、0.8的聚类大小分布
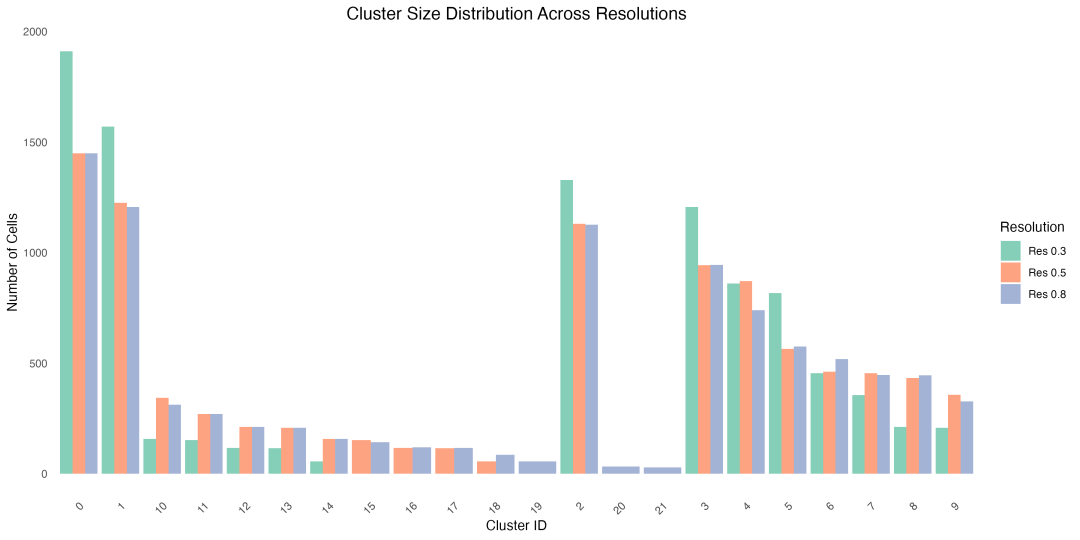
图 4 不同分辨率下聚类大小分布:展示了分辨率0.3、0.5、0.8下各聚类的细胞数量分布,帮助评估聚类的稳定性。
稳定性分析显示,主要的聚类在不同分辨率下都保持了相对稳定的大小,这表明这些聚类代表了真实的细胞群体而非算法的人工产物。随着分辨率增加,一些大聚类会分化成多个小聚类,这是正常的细分过程。
聚类质量的最终评估需要检查每个聚类的基本特征,包括细胞数量、转录组质量指标等。
# 计算每个聚类的统计信息
cluster_stats <- scRNA@meta.data %>%
group_by(RNA_snn_res.0.5) %>%
summarise(
n_cells = n(),
mean_nCount = mean(nCount_RNA),
mean_nFeature = mean(nFeature_RNA),
mean_mt = mean(percent.mt)
)
# 生成质量评估热图
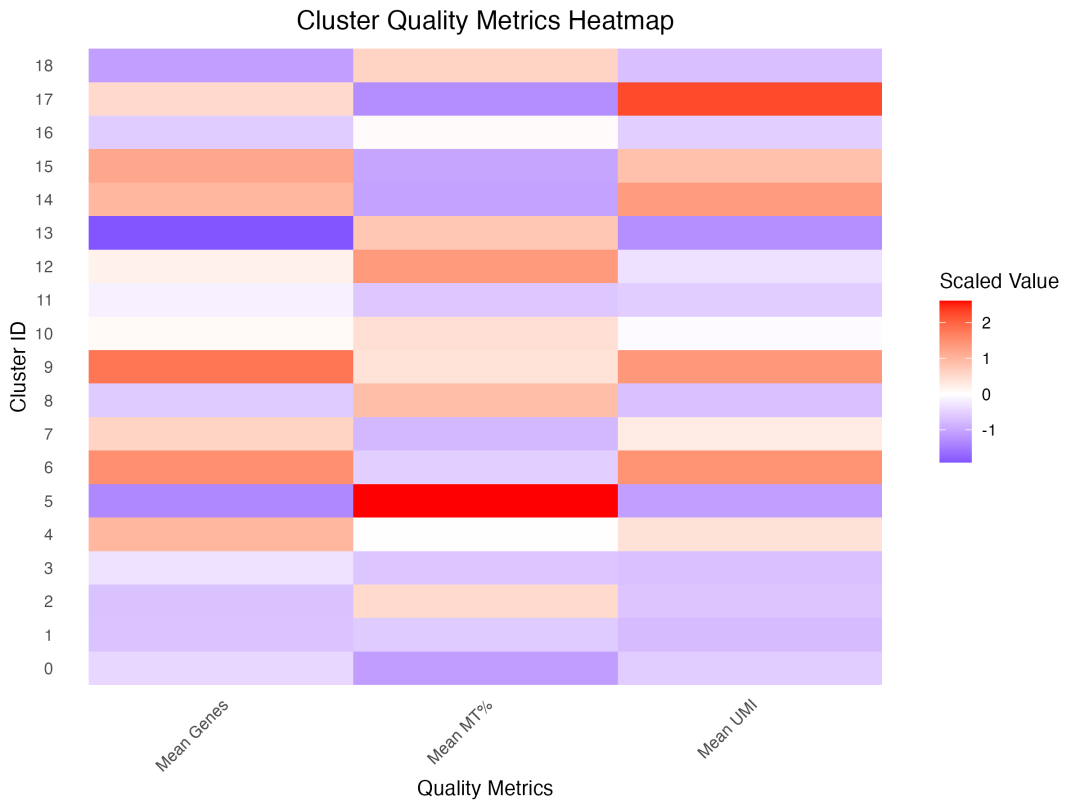
图 5 聚类质量指标热图:展示了每个聚类在UMI计数、检测基因数和线粒体基因百分比等质量指标上的表现。
质量评估结果显示,大部分聚类的质控指标都在合理范围内,不同聚类间的差异主要反映了细胞类型的生物学特性而非技术性问题。例如,某些聚类具有更高的UMI计数和基因数,这可能对应于转录更活跃的细胞类型;某些聚类的线粒体基因比例略高,这可能反映了不同细胞类型的代谢特征。
聚类分析的最终目标是为后续的细胞类型注释和功能分析提供基础。高质量的聚类结果应该具有以下特征:聚类数量生物学合理、聚类大小适中(既不过大也不过小)、聚类间有清晰的转录组差异、聚类内的细胞具有相似的生物学特征。
总结
-
图构建是基础 :基于SNN的邻居图构建为稳定聚类提供了坚实基础 -
分辨率选择很关键 :需要结合clustree分析和生物学知识选择最优分辨率 -
多角度验证必须 :通过样本分布、稳定性分析、质量评估全面验证聚类结果 -
生物学意义优先 :聚类结果必须符合生物学预期和实验设计逻辑
聚类分析将复杂的单细胞数据转化为有意义的细胞群体,为理解组织的细胞异质性和功能多样性提供了重要基础。正确的聚类分析不仅能够准确识别已知的细胞类型,还可能发现新的细胞亚型和过渡状态,为生物学发现开辟新的方向。