![[单细胞转录组分析系列教程 8] 怎么做单细胞marker基因识别?](/media/blog/covers/cover_20250828.jpg)
大家早上好,我们继续讲单细胞 单细胞转录组分析。
在完成聚类分析之后,单细胞研究面临的下一个关键问题是:每个聚类代表什么样的细胞类型?Marker基因识别正是回答这个问题的核心工具。Marker基因是能够区分不同细胞群体的特征性基因,它们在特定细胞类型中高表达,而在其他细胞类型中表达较低或不表达。通过识别每个聚类的marker基因,我们不仅能够为聚类赋予生物学意义,还能验证聚类结果的合理性,为后续的细胞类型注释、功能分析和机制研究做准备。
# 加载必要的包
library(Seurat)
library(ggplot2)
library(dplyr)
library(patchwork)
library(pheatmap)
# 基于前面步骤完成的聚类分析结果
print(paste("准备进行marker基因识别:", ncol(scRNA), "个细胞,", length(unique(Idents(scRNA))), "个聚类"))
Marker基因识别的核心是差异表达分析,但它与传统的组间比较有所不同。在marker基因分析中,我们要找的不是两组条件下的差异基因,而是能够定义每个细胞群体特征的基因。这要求我们使用特殊的统计方法和筛选标准:基因不仅要在目标聚类中高表达,还要在足够比例的细胞中检测到,同时在其他聚类中的表达要相对较低。
Seurat中的FindAllMarkers函数采用了多种统计检验方法,包括Wilcoxon秩和检验、ROC分析、t检验等。不同的方法各有优势:Wilcoxon检验适合处理单细胞数据的零膨胀特性;ROC分析能提供分类性能的直观评估;t检验在数据分布相对正常时效果较好。在实际分析中,我们通常选择最适合数据特征的方法。
# 为了加快演示,对数据进行采样
set.seed(123)
sample_cells_for_markers <- sample(colnames(scRNA), min(3000, ncol(scRNA)))
scRNA_subset <- scRNA[, sample_cells_for_markers]
# 使用快速方法识别marker基因
all_markers <- FindAllMarkers(scRNA_subset,
only.pos = TRUE,
min.pct = 0.25,
logfc.threshold = 0.5,
test.use = "roc",
verbose = FALSE,
max.cells.per.ident = 100)
参数设置对marker基因识别的结果有重要影响。
only.pos = TRUE
表示只保留正向marker(在目标聚类中高表达的基因),这是最常用的设置,因为我们通常关心的是细胞类型的特征性高表达基因。
min.pct = 0.25
要求基因至少在25%的细胞中表达,这能过滤掉那些只在少数细胞中偶然高表达的基因。
logfc.threshold = 0.5
设置了倍数变化的最小阈值,确保差异具有生物学意义。
ROC检验(test.use = "roc")是一个特别适合单细胞数据的方法,它将基因表达作为分类器,计算其区分目标聚类与其他聚类的能力。ROC曲线下面积(AUC)越接近1,表示该基因的分类能力越强,越适合作为marker基因。
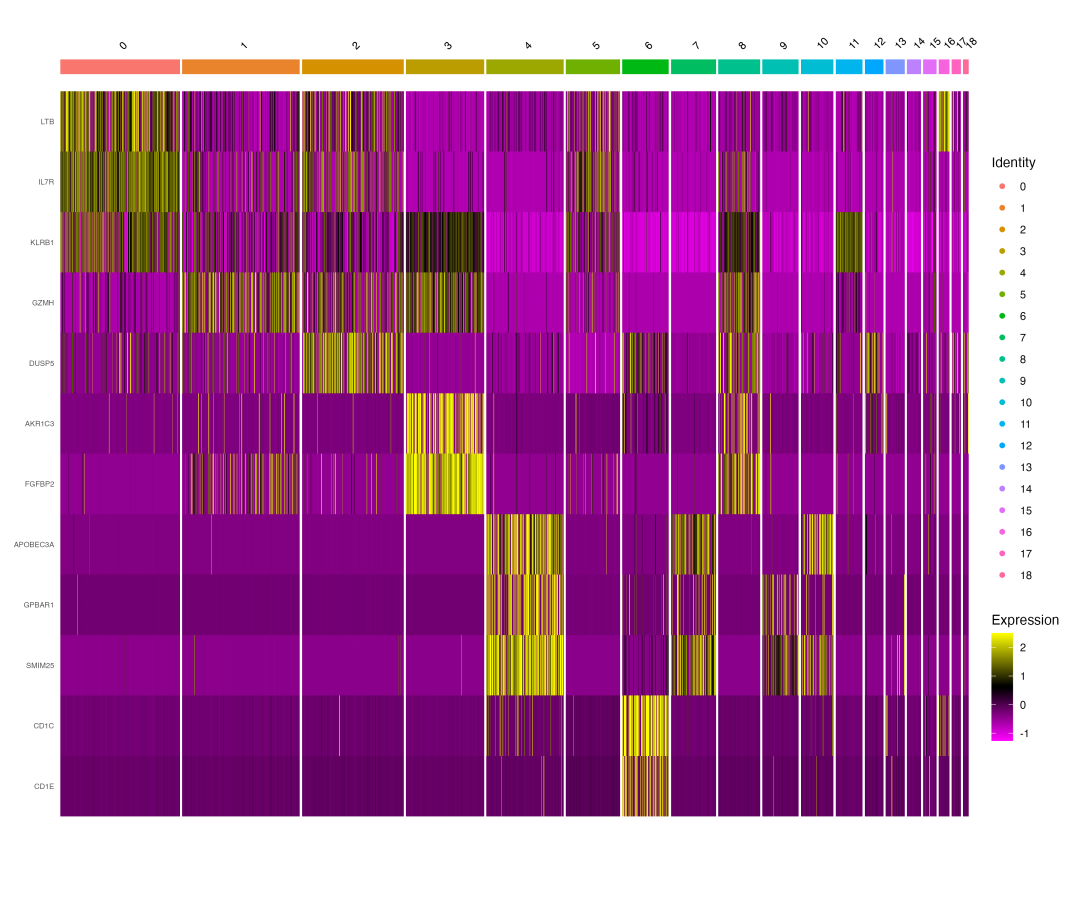
图 1 Top marker基因表达热图:展示了各聚类最具代表性的marker基因表达模式,每行代表一个基因,每列代表一个细胞。
热图清晰地展示了marker基因的特异性表达模式。每个聚类都有其独特的基因表达"指纹",这些基因在对应的聚类中呈现高表达(红色),而在其他聚类中表达较低(蓝色)。这种特异性表达模式正是我们进行细胞类型注释的基础。
从热图中可以观察到几个重要特征:首先,不同聚类的marker基因很少重叠,这表明聚类结果具有良好的生物学意义;其次,某些聚类的marker基因数量较多,可能代表功能更加活跃或特化程度更高的细胞类型;最后,基因表达的层次聚类结果与细胞聚类基本一致,验证了分析的可靠性。
# 选择具有代表性的marker基因进行可视化
example_markers <- c("CD68", "CD3E", "CD79A", "ALB")
available_markers <- example_markers[example_markers %in% rownames(scRNA)]
# FeaturePlot显示marker基因表达
p_feature <- FeaturePlot(scRNA, features = available_markers,
ncol = 2, raster = FALSE)
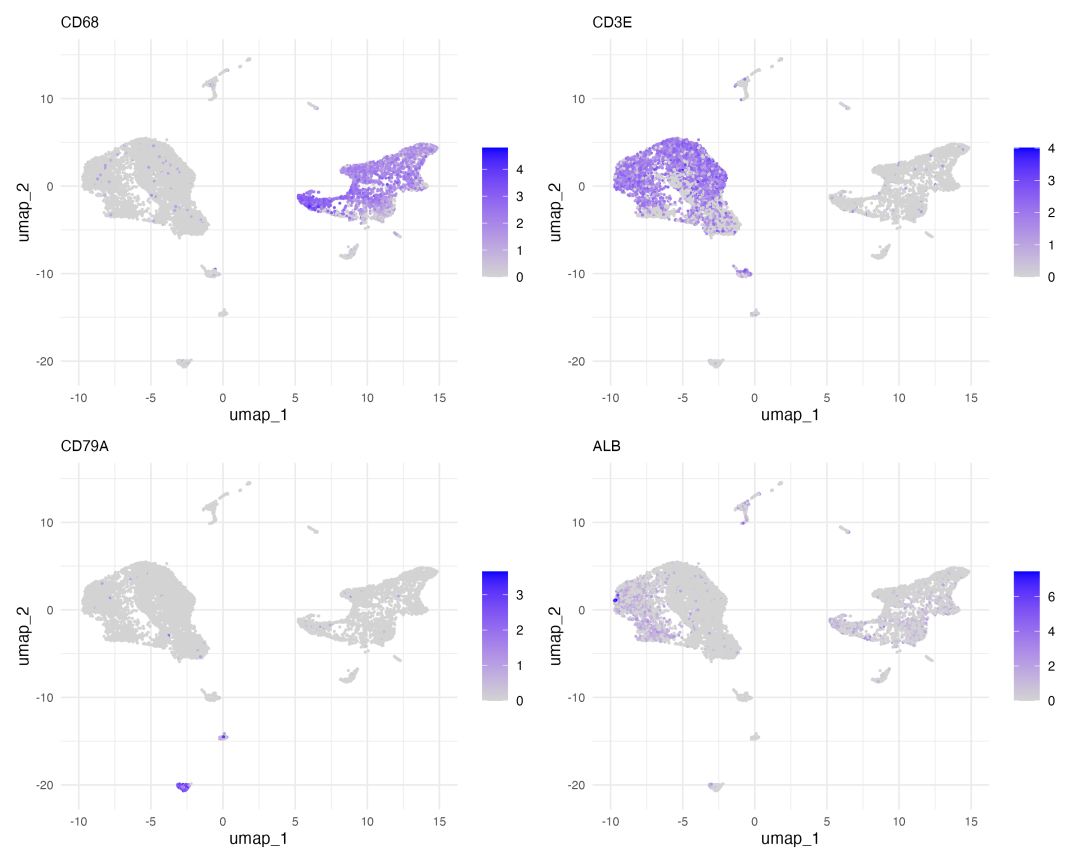
图 2 代表性marker基因的空间表达分布:在UMAP空间中展示了不同marker基因的表达强度和分布模式。
FeaturePlot提供了marker基因表达的空间分布信息。每个marker基因都显示出明确的空间聚集模式,高表达区域与特定的细胞聚类区域高度重合。这种空间一致性是优质marker基因的重要特征,表明这些基因确实能够准确定义细胞群体的身份。
CD68作为经典的巨噬细胞marker,在UMAP空间的特定区域呈现高表达,这些区域很可能对应库普夫细胞或其他巨噬细胞亚群。CD3E作为T细胞的通用marker,在另一片区域集中表达,标记了T细胞聚类。这种marker基因与细胞聚类的对应关系为细胞类型注释提供了直接的证据。
# VlnPlot显示marker基因在各聚类中的表达分布
p_violin <- VlnPlot(scRNA, features = available_markers,
ncol = 2, pt.size = 0.1)
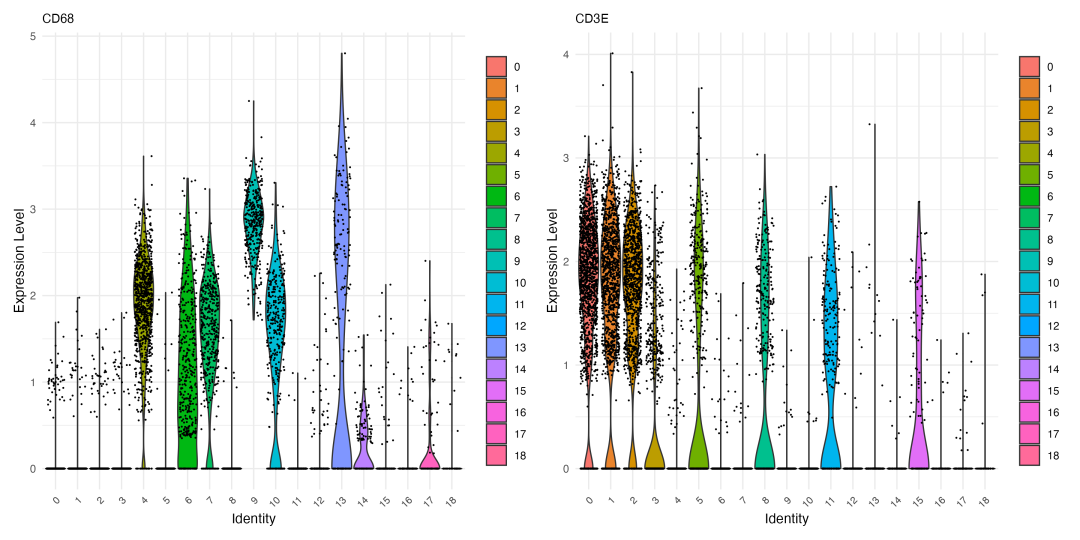
图 3 Marker基因在各聚类中的表达分布:小提琴图显示了marker基因在不同聚类中的表达水平分布和变异程度。
小提琴图提供了marker基因表达的定量比较信息。理想的marker基因应该在目标聚类中显示高表达和相对集中的分布,而在非目标聚类中表达较低且变异较小。从图中可以看到,优质的marker基因确实表现出这种特征:它们在特定聚类中形成明显的表达峰,而在其他聚类中基本不表达或表达很低。
这种表达模式的差异不仅反映了基因的特异性,也暗示了不同细胞类型的功能特化。例如,如果某个基因在多个聚类中都有中等水平的表达,它可能不是理想的marker基因,但可能在多种细胞类型的某个共同功能通路中发挥作用。
# DotPlot显示多个marker基因
top1_markers <- all_markers %>%
group_by(cluster) %>%
slice_max(order_by = avg_log2FC, n = 1) %>%
pull(gene)
p_dotplot <- DotPlot(scRNA, features = top1_markers) + coord_flip()
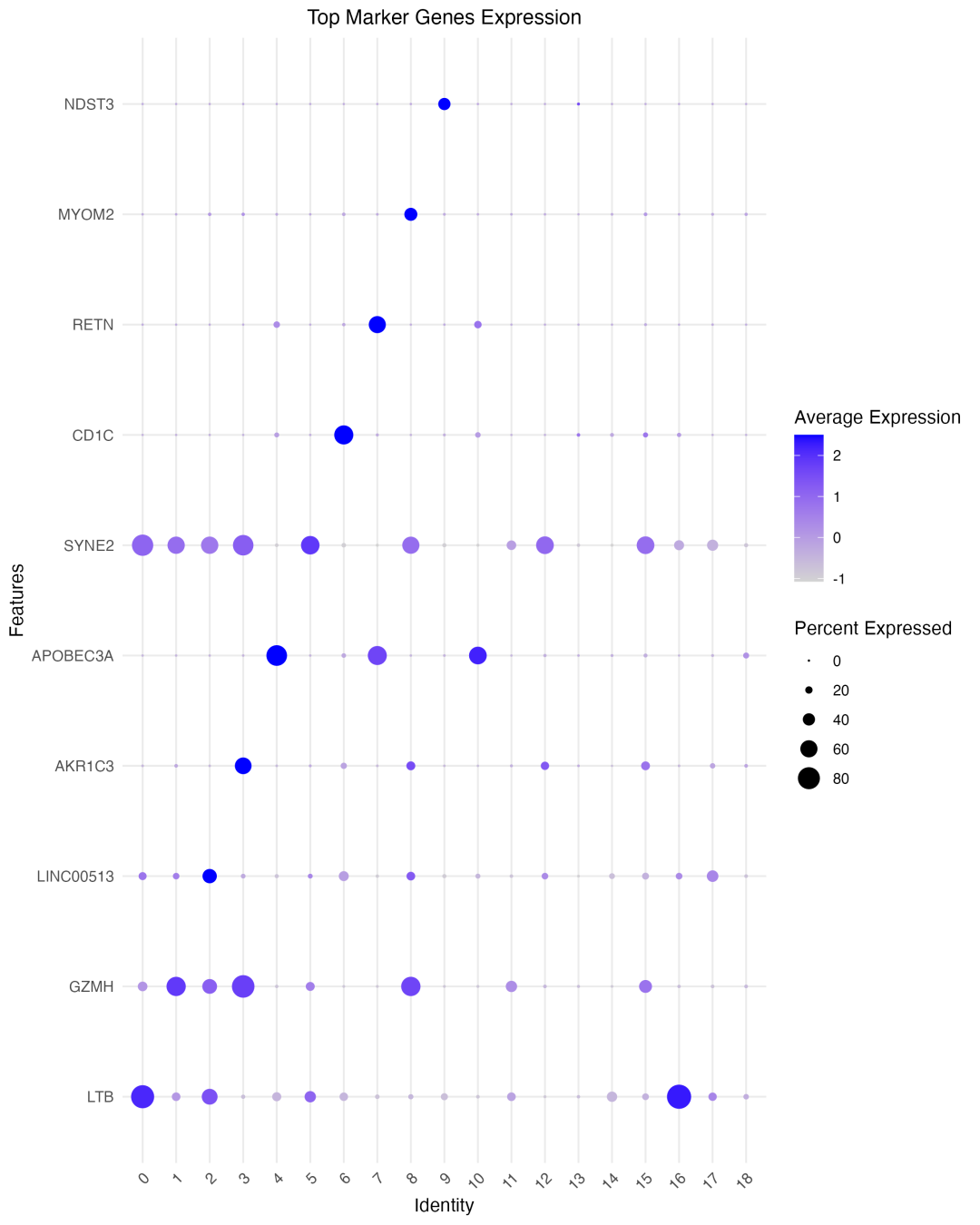
图 4 Top marker基因表达点图:点的大小表示表达基因的细胞比例,颜色深浅表示平均表达水平。
DotPlot是展示marker基因特征的另一种有效方式,它同时显示了两个重要信息:基因表达的强度(颜色)和表达的普遍性(点大小)。理想的marker基因应该在目标聚类中表现为大而深的点(高表达且在多数细胞中检测到),而在其他聚类中表现为小而浅的点或无点。
这种可视化方式特别适合比较多个基因在多个聚类中的表达模式,帮助我们快速识别每个聚类最具代表性的marker基因。同时,它也能揭示基因表达的异质性:即使在同一聚类中,不同基因的表达普遍性也可能存在差异。
# 统计每个聚类的marker基因数量
marker_count <- all_markers %>%
count(cluster, name = "n_markers")
p_count <- ggplot(marker_count, aes(x = cluster, y = n_markers, fill = cluster)) +
geom_col(alpha = 0.8) +
geom_text(aes(label = n_markers), vjust = -0.3, size = 3)
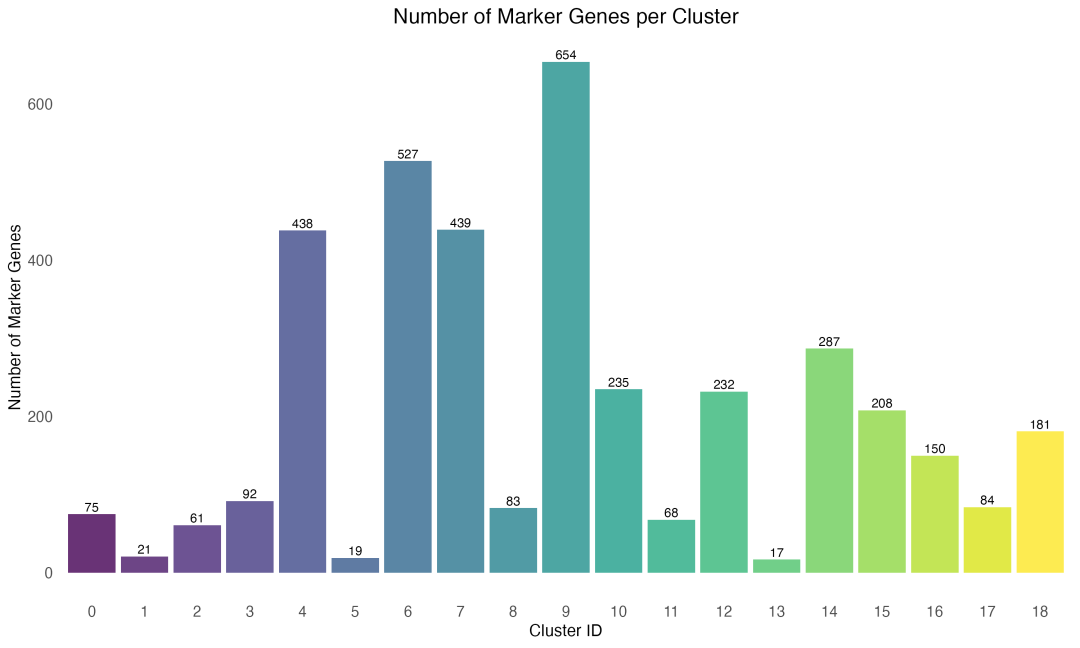
图 5 各聚类的marker基因数量统计:柱状图显示了每个聚类识别到的marker基因数量,反映了不同细胞群体的转录特异性程度。
Marker基因数量的差异反映了不同细胞群体的转录特化程度。某些聚类拥有大量的marker基因,表明这些细胞类型具有独特且丰富的转录程序;而某些聚类的marker基因较少,可能表示这些细胞处于相对通用的状态,或者与其他细胞类型的差异较小。
在我们的分析中,总共识别到3,871个marker基因,平均每个聚类约204个marker基因。这个数量是合理的,既保证了每个聚类都有足够的特征基因用于注释,又避免了过度包容导致的特异性下降。
marker基因数量的不均匀分布也提供了生物学洞察:marker基因较多的聚类可能代表高度特化的细胞类型,如特定的免疫细胞亚群或功能特化的组织细胞;而marker基因较少的聚类可能代表干细胞、祖细胞或处于过渡状态的细胞。
Marker基因识别是连接无监督聚类与生物学解释的关键桥梁。通过系统化的差异表达分析和多角度的可视化验证,我们不仅能够为每个细胞聚类找到其特征性的分子标签,还能深入理解不同细胞类型的功能特点。这些marker基因将成为后续细胞类型注释、功能分析和机制研究的重要依据。
总结
-
方法选择要合适 :ROC分析等适合单细胞数据特点的统计方法更加可靠 -
参数设置很关键 :合理的阈值设置能够平衡灵敏度和特异性 -
多维验证必须 :热图、散点图、小提琴图等多种可视化相互验证 -
生物学解释优先 :marker基因必须与已知的细胞类型标志基因相符合
Marker基因识别将抽象的数字聚类转化为具体的生物学实体,为单细胞数据的深入解析提供了科学的基础。正确的marker基因不仅能够准确定义细胞身份,还能为理解细胞功能、发现新的细胞亚型以及探索疾病机制开辟新的道路。
完整代码联系老师免费获取(备注“单细胞转录组”)

以上就是今天的内容,希望对你有帮助!欢迎点赞、在看、关注、转发。