大家早上好呀,在转录组差异表达分析中,你可能发现大多数主流工具如DESeq2、edgeR都要求输入原始的read count数据,而不是FPKM、TPM等标准化后的数据。这背后有什么统计学原理吗?
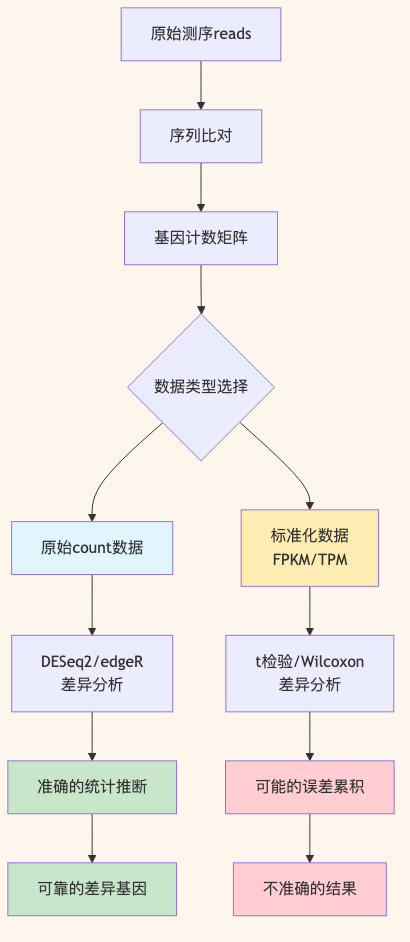
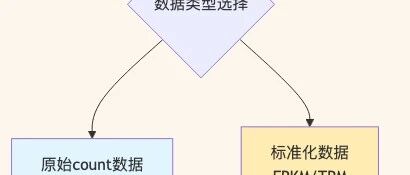
图 1 差异表达分析中数据类型选择的决策流程:原始count数据能够保留重要的统计信息。
原始count数据的统计学优势
RNA-seq产生的read count数据本质上遵循 负二项分布 (negative binomial distribution),这种离散型分布能够很好地描述测序计数的随机性。DESeq2和edgeR正是基于这一分布假设构建统计模型,能够准确估计基因表达的均值和方差关系。
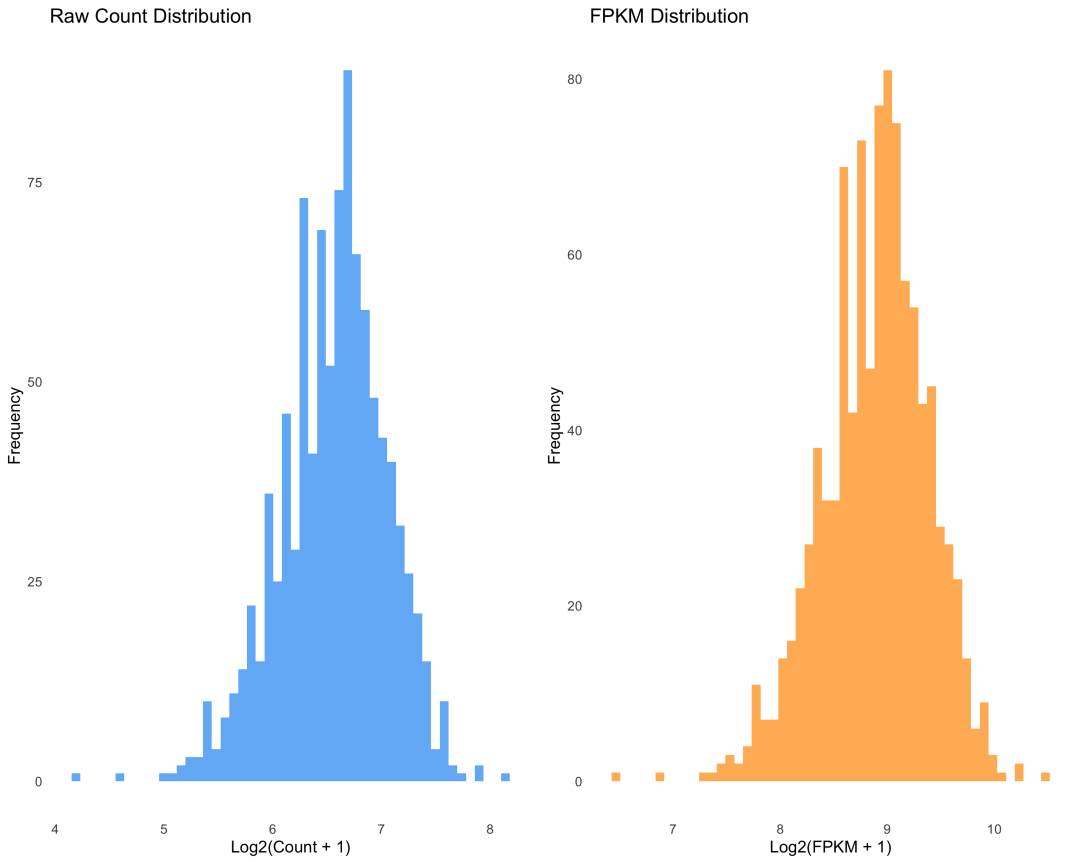
图 2 原始count数据与FPKM数据的分布差异:count数据保持了天然的计数特性。
当我们对count数据进行FPKM/TPM标准化时,虽然消除了测序深度和基因长度的影响,但也破坏了数据的原始分布特性。标准化后的数据不再遵循负二项分布,而是变成了连续型数据,传统的t检验等方法可能不再适用。
方差建模的关键差异
count数据具有一个重要特性: 方差与均值存在特定的函数关系 。在负二项分布中,方差通常大于均值(过度离散),且这种关系在不同表达水平的基因中表现一致。
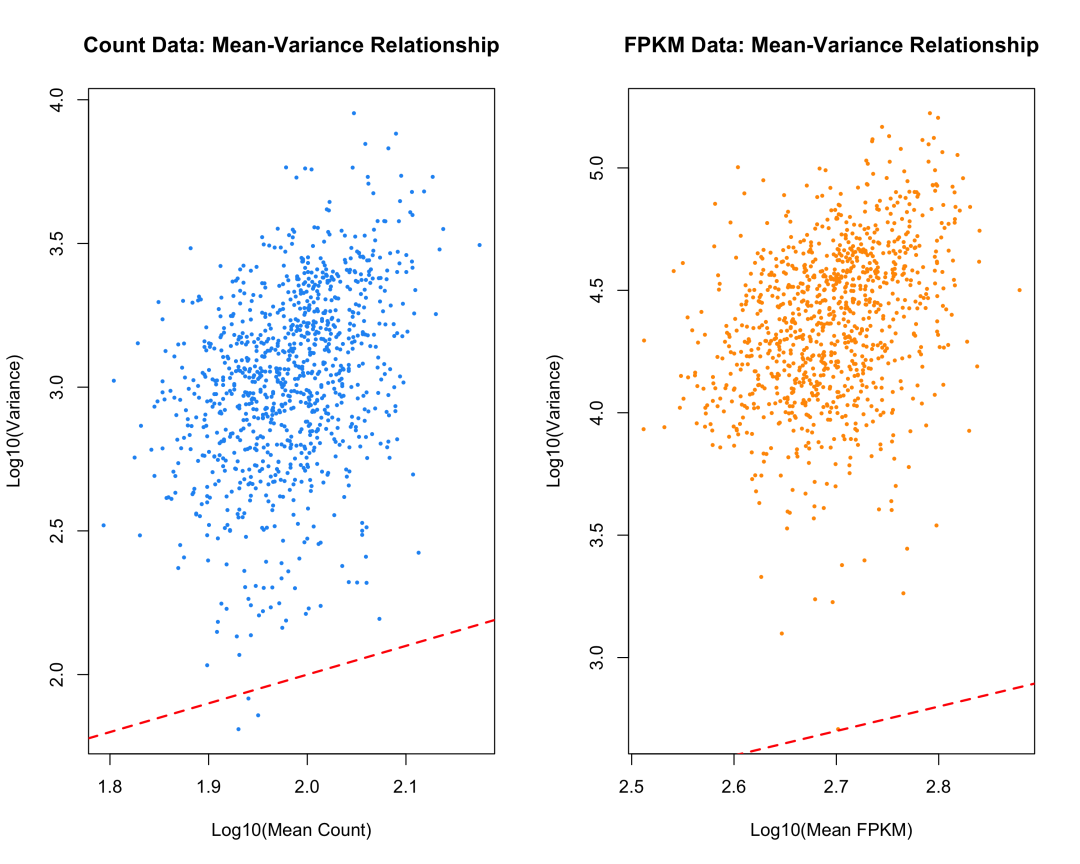
图 3 count数据与FPKM数据的均值-方差关系:count数据展现出更稳定的统计关系。
DESeq2通过建模这种方差-均值关系,能够:
-
-
为低表达基因"借用"信息,提高统计检验的稳定性 -
精确估计离散度参数,控制假阳性率 -
处理生物学重复间的真实变异
相比之下,FPKM等标准化数据失去了这种天然的统计结构,难以进行可靠的方差估计。
现代差异分析的最佳实践
基于原始count数据的差异分析流程已成为金标准:
-
-
保留完整统计信息 :count数据承载了测序实验的全部随机性信息 -
内置标准化 :DESeq2等工具内部使用median-of-ratios等稳健方法处理样本间差异 -
精确假设检验 :基于负二项分布的Wald检验或似然比检验更加准确
最后
选择原始count数据进行差异表达分析,本质上是尊重数据的统计本质。这种做法不仅保证了分析结果的可靠性,也为后续的功能富集分析奠定了坚实基础。在追求生物学发现的路上,统计学的严谨性永远是第一步。
以上就是今天的内容,希望对你有帮助!欢迎点赞、在看、关注、转发。