今天,我们讲一下如何进行KEGG富集分析,KEGG富集分析其实是一个非常实用的生物信息学分析方法,用来找出一组基因与哪些生物学通路(Pathways)有关。通路指的是细胞或生物体内一系列相互作用的分子,如酶、信号分子、受体等,它们一起合作完成某些特定的生物学功能。
在开始进行KEGG富集分析之前,确保你已经完成了差异表达分析(
【GEO数据挖掘系列】(2) 差异表达分析及可视化
),并得到了一个包含差异表达基因的
sign_diff_expr_results.txt
文件(过滤条件是p_adj=0.05 log2fc=1)。p_adj是校正后的p值表示结果显著,log2fc=1表示差异变化倍数为2。(对p_adj和log2fc的详细解释可以看这篇文章)(
基因差异表达分析中,为什么选择的是log2FC和padj值来筛选关键基因,而不是log2FC和p值
)
读取差异表达基因
读取差异分析后得到差异表达基因文件名
sign_diff_expr_results.txt
,具体参考这篇文章(
【GEO数据挖掘系列】(2) 差异表达分析及可视化
)
# 安装必要的R包
if (!require("clusterProfiler")) install.packages("clusterProfiler")
if (!require("org.Hs.eg.db")) install.packages("org.Hs.eg.db")
if (!require("ggplot2")) install.packages("ggplot2")
if (!require("enrichplot")) install.packages("enrichplot")
# 加载R包
library(clusterProfiler)
library(org.Hs.eg.db)
library(ggplot2)
library(enrichplot)
# 读取差异表达基因文件
diff_genes <- read.table("sign_diff_expr_results.txt", row.names=1, header=T)
KEGG富集分析
同样地,我们进行KEGG富集分析。
# 提取基因ID
gene_list <- rownames(diff_genes)
genes <- unique(gene_list) # 提取输入文件中第一列的唯一基因名称
entrez_ids <- mget(genes, org.Hs.egSYMBOL2EG, ifnotfound = NA) # 根据基因名称获取对应的Entrez ID
entrez_ids <- as.character(entrez_ids) # 将基因ID转换为字符向量类型
gene_group_diff_matrix <- data.frame(genes, entrez_id = entrez_ids) # 创建包含基因名称和Entrez ID的数据框
gene <- entrez_ids[entrez_ids != "NA"] # 从Entrez ID中去除NA值,得到有效的基因ID
# KEGG富集分析
kegg_enrich <- enrichKEGG(gene = gene,
organism = 'hsa',
pAdjustMethod = "BH",
pvalueCutoff = 0.05,
qvalueCutoff = 0.05)
KEGG <- as.data.frame(kegg_enrich) # 将富集结果转换为数据框格式
KEGG$geneID <- as.character(sapply(KEGG$geneID, function(x) paste(gene_group_diff_matrix$genes[match(strsplit(x, "/")[[1]], as.character(gene_group_diff_matrix$entrez_id))], collapse = "/"))) # 将geneID转换为基因名称,并加入到KEGG结果中
# 查看KEGG富集分析结果
head(KEGG)
write.table(KEGG, file = "kegg_enrichment.tsv", sep = "\t", quote = FALSE, row.names = FALSE) # 将KEGG结果写入名为kegg.tsv的文件中,以制表符分隔,不包含行名
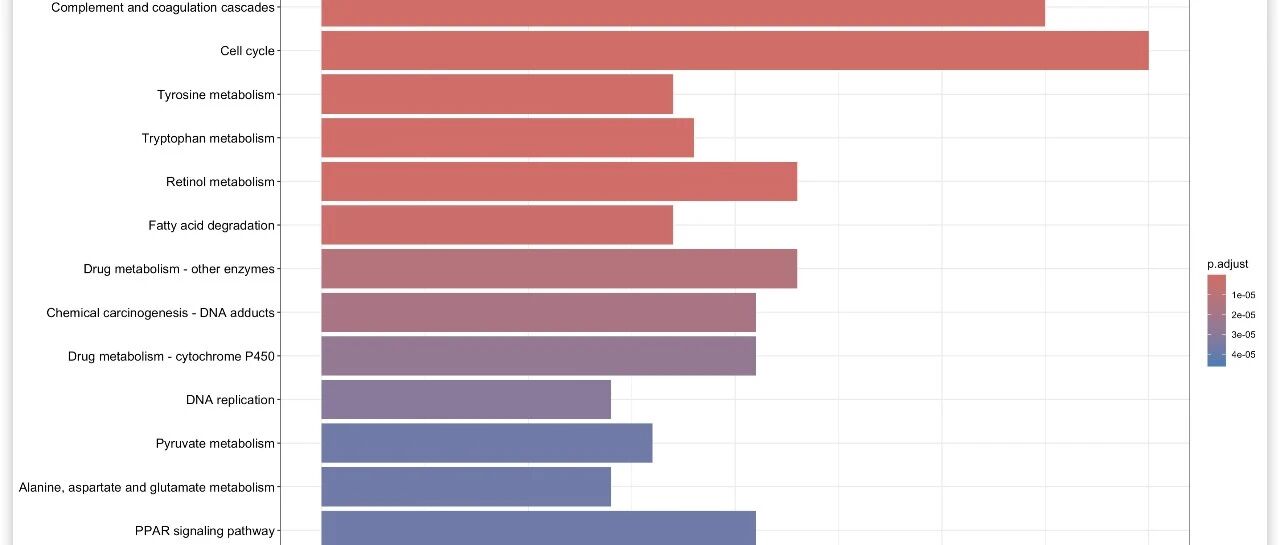
KEGG富集分析结果文件
kegg_enrichment.tsv
如下
接着我们对KEGG富集分析结果文件进行可视化
绘制条形图
条形图是展示富集分析结果的常用方式。我们可以使用
barplot
函数来绘制KEGG富集分析的条形图。
# 绘制KEGG富集分析条形图
barplot(kegg_enrich, drop = TRUE, showCategory = 15, label_format = 60)
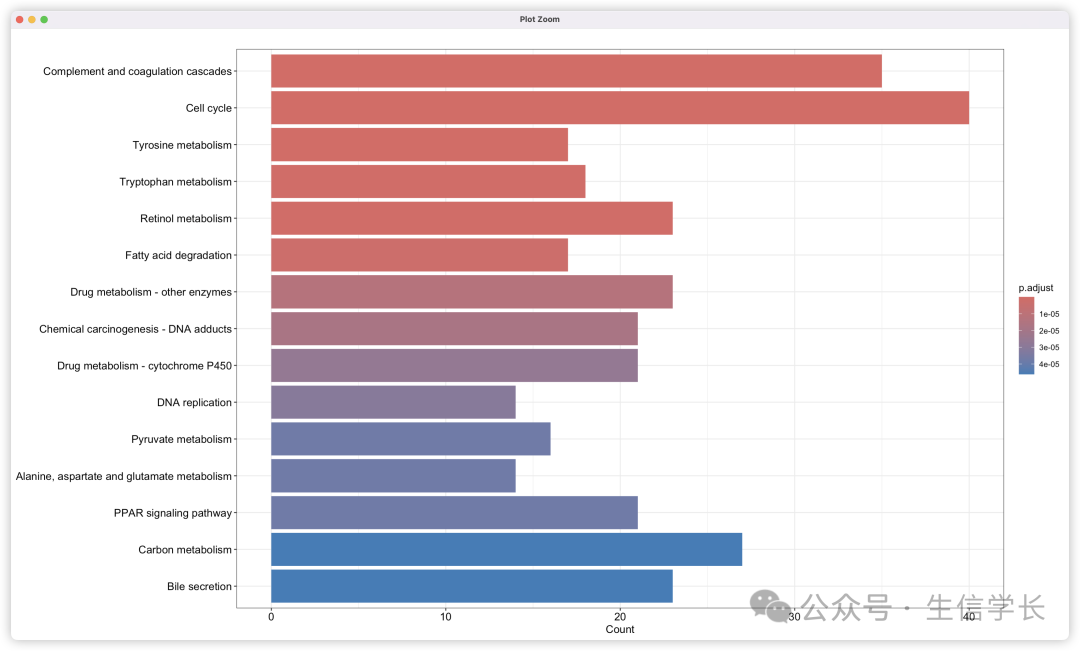
KEGG富集分析条形图的解读可以参考GO富集分析的条形图( 【GEO数据挖掘系列】(4) GO富集分析 )
绘制气泡图
气泡图是另一种展示富集分析结果的方式,它可以同时展示富集项的p值和基因数量。
# 绘制KEGG富集分析气泡图
dotplot(kegg_enrich, showCategory = 15, orderBy = "GeneRatio", label_format = 60) 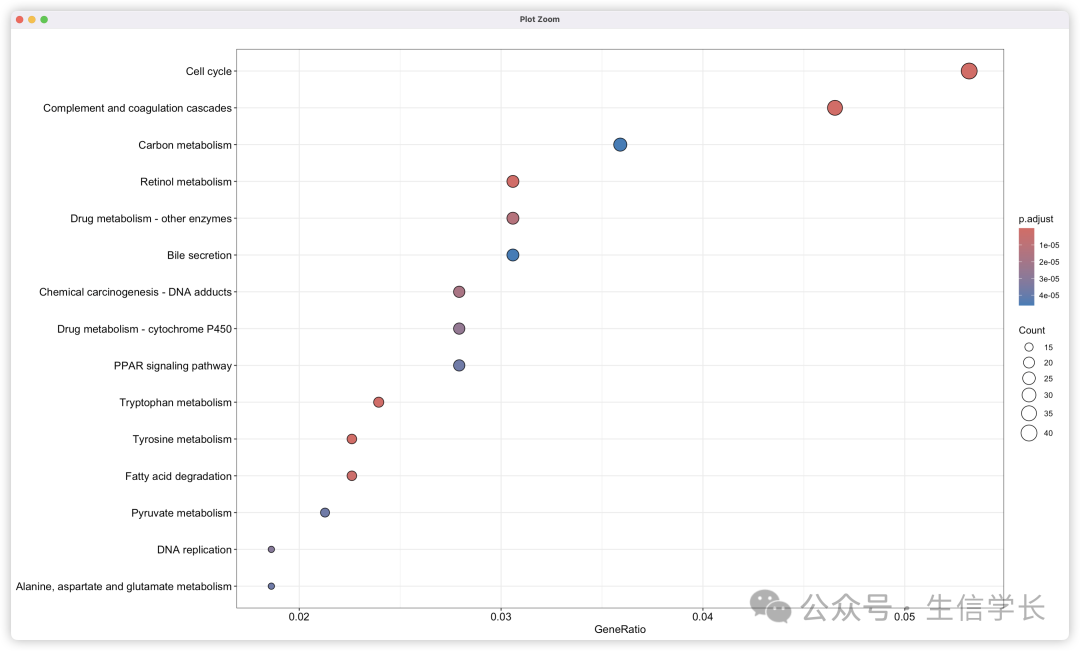
KEGG富集分析气泡图的解读可以参考GO富集分析的气泡图( 【GEO数据挖掘系列】(4) GO富集分析 )
以上就是KEGG富集分析的内容,我们讲了KEGG富集分析,并绘制条形图和气泡图图。这些图表可以帮助我们直观地理解差异表达基因在通路中的功能。例如,KEGG富集分析可能显示这些基因在“癌症通路”或“代谢通路”中显著富集。
最后补充一点,KEGG富集分析和GO富集分析通常是一起进行的,它们分别从不同的角度对基因集的功能进行解释。例如我们正在研究某种疾病(如癌症)相关的基因。通过GO富集分析,你可能发现这些基因显著富集于“细胞增殖”和“细胞凋亡”等生物学过程。而通过KEGG富集分析,你可能进一步发现这些基因富集于“癌症通路”和“细胞周期通路”。这样就更可能说明,这些基因不仅在基本的生物学过程上有显著作用,它们还参与了特定的生物学通路,可能是通过这些通路来调控癌症的发生。
写作不易,欢迎关注