大家早上好!今天跟大家讲一下 GSVA Hallmark 基因集变异分析 !
相信很多小伙伴都听说过通路分析,但是今天要介绍的 GSVA 方法有什么特别的呢?简单来说,传统的 GO、KEGG 富集分析需要你先找到差异基因,然后看这些基因富集在哪些通路上。而 GSVA 直接给每个样本的每个通路打分 ,不需要提前筛选基因,这样我们就能更全面地比较不同样本之间的通路活性差异!
今天我们使用的是 MSigDB Hallmark 基因集 ,这套基因集包含了 50 个关键生物学过程,比如脂肪生成、免疫反应、DNA 修复等等,每个都有明确的生物学意义,结果特别好解释
第一步:数据加载和初步探索
首先,我们来看看今天要分析的数据:
# 加载必需的R包
library(GSVA) # GSVA分析核心包
library(GSEABase) # 读取GMT基因集文件
library(limma) # 差异分析
library(ggplot2) # 绘图神器
library(pheatmap) # 热图专家
library(dplyr) # 数据处理
# 读取表达矩阵和Hallmark基因集
expr_matrix <- read.table("expression_matrix.tsv", header = TRUE, row.names = 1, sep = "\t")
gene_sets <- getGmt("h.all.v2025.1.Hs.symbols.gmt")
print(sprintf("表达矩阵: %d 基因 x %d 样本", nrow(expr_matrix), ncol(expr_matrix)))
print(sprintf("Hallmark基因集数量: %d", length(gene_sets)))
运行结果:
表达矩阵: 59427 基因 x 448 样本 Hallmark基因集数量: 50 我们有近 6 万个基因在 448 个样本中的表达数据,还有 50 个精选的 Hallmark 通路!这些 Hallmark 通路包括:
-
ADIPOGENESIS(脂肪生成) -
ALLOGRAFT_REJECTION(同种异体排斥) -
ANDROGEN_RESPONSE(雄激素反应) -
ANGIOGENESIS(血管生成) -
P53_PATHWAY(p53 通路)
第二步:数据预处理
在跑 GSVA 之前,我们需要对数据做一些必要的预处理:
# 检查数据分布
data_range <- range(expr_matrix, na.rm = TRUE)
print(sprintf("表达值范围: %.2f 到 %.2f", data_range[1], data_range[2]))
# 如果是原始counts数据,进行log2转换
if(max(expr_matrix, na.rm = TRUE) > 50) {
print("检测到原始counts数据,正在进行log2转换...")
expr_matrix <- log2(expr_matrix + 1)
new_range <- range(expr_matrix, na.rm = TRUE)
print(sprintf("转换后: %.2f 到 %.2f", new_range[1], new_range[2]))
}
# 过滤低表达基因(在至少20%样本中有表达)
keep_genes <- rowSums(expr_matrix > 0) >= ncol(expr_matrix) * 0.2
expr_matrix <- expr_matrix[keep_genes, ]
print(sprintf("过滤后保留基因: %d", nrow(expr_matrix)))
运行结果:
表达值范围: 0.00 到 173492.24 检测到原始counts数据,正在进行log2转换... 转换后: 0.00 到 17.40 过滤后保留基因: 47202 为什么要这样处理?
-
Log2 转换 :原始 counts 数据分布很不均匀,log 转换后能让数据更符合正态分布 -
过滤低表达基因 :那些在大部分样本中都不表达的基因其实没什么意义,去掉能减少噪音
第三步:样本分组
为了后续的差异分析,我们需要给样本分组。这里我用一个简单但有效的策略:
# 基于样本整体表达水平分组
sample_median_expr <- apply(expr_matrix, 2, median)
median_cutoff <- median(sample_median_expr)
sample_groups <- data.frame(
Sample = colnames(expr_matrix),
Group = ifelse(sample_median_expr > median_cutoff, "High_Expr", "Low_Expr"),
stringsAsFactors = FALSE
)
table(sample_groups$Group)
运行结果:
High_Expr Low_Expr 224 224 我们得到了两个平衡的组:高表达组和低表达组,每组 224 个样本。在实际研究中,你可以根据疾病状态、治疗条件、时间点等进行分组。
第四步:GSVA 分析 - 关键步骤!
现在到了最核心的步骤——运行 GSVA 分析:
# 运行GSVA分析
print("开始GSVA分析,请稍候...")
gsva_results <- gsva(
as.matrix(expr_matrix),
gene_sets,
method = "gsva", # 使用GSVA方法
kcdf = "Gaussian", # 核密度估计
min.sz = 10, # 最小基因集大小
max.sz = 500, # 最大基因集大小
verbose = FALSE
)
print(sprintf("GSVA分析完成!分析了 %d 个Hallmark通路", nrow(gsva_results)))
运行结果:
GSVA分析完成!分析了 50 个Hallmark通路 我们现在得到了一个 50×448 的富集分数矩阵,每行是一个通路,每列是一个样本,矩阵中的数值就是 GSVA 富集分数 :
-
正值 :该通路在该样本中被激活 -
负值 :该通路在该样本中被抑制
第五步:结果可视化 - GSVA 分数分布
让我们先来看看 GSVA 分数的整体分布情况:
# 生成多面板分布图
png("figure1_hallmark_distribution.png", width = 1200, height = 800, res = 150)
par(mfrow = c(2, 2), mar = c(4.5, 4.5, 3, 1.5))
# 分数分布直方图
hist(as.vector(gsva_results),
main = "Hallmark GSVA Score Distribution",
xlab = "GSVA Enrichment Score",
ylab = "Frequency",
breaks = 60,
col = alpha("steelblue", 0.7),
border = "white")
abline(v = 0, col = "red", lwd = 3, lty = 2)
# 其他3个子图...
dev.off()
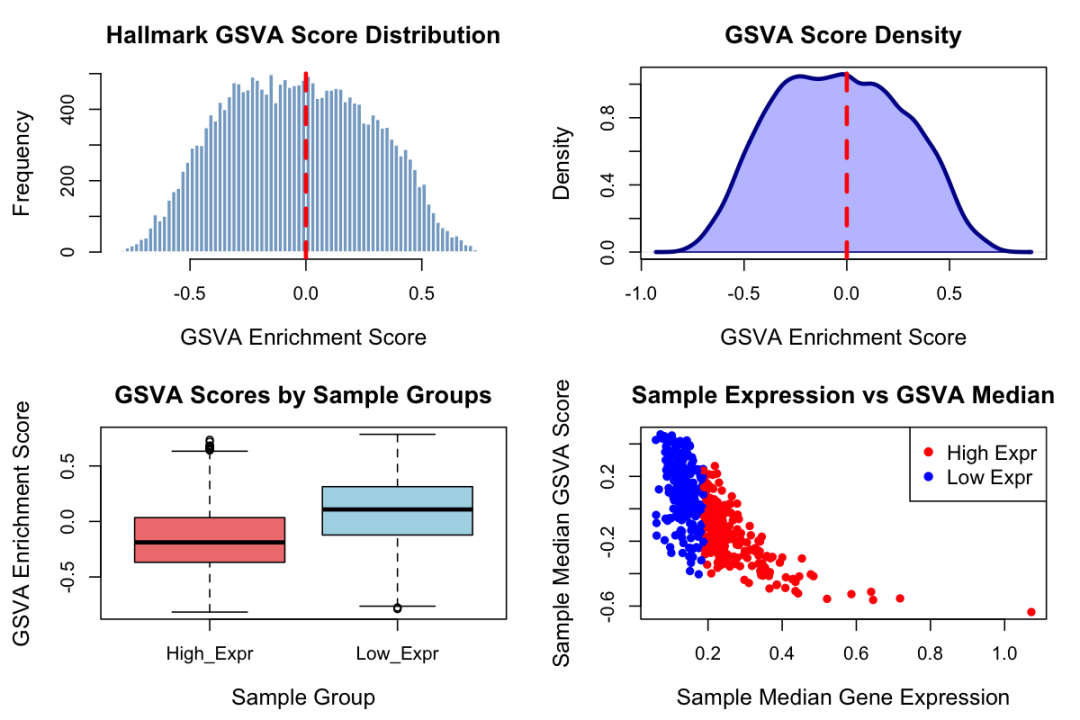
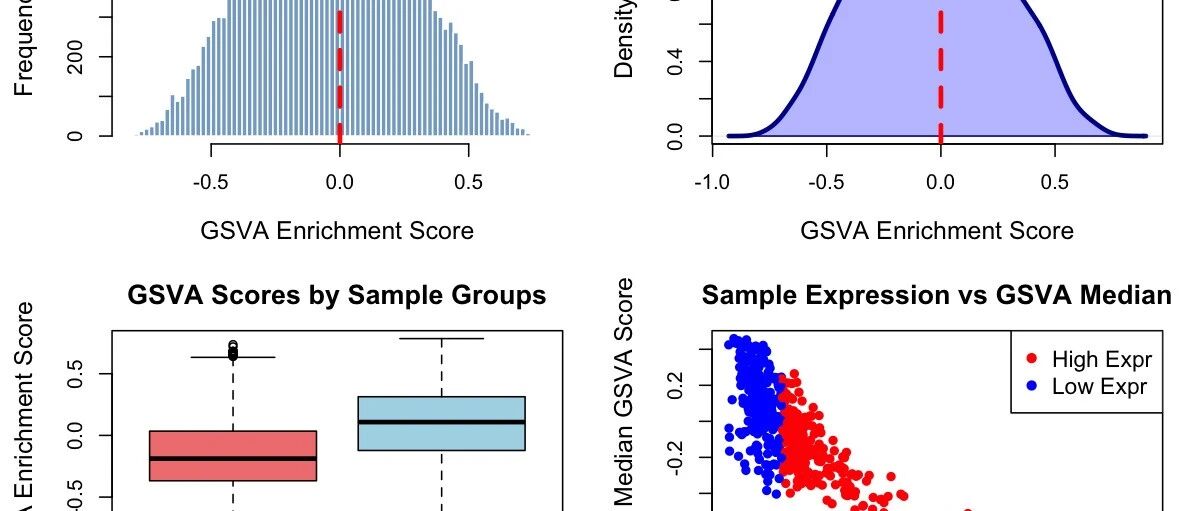
结果解读:
-
左上图 :GSVA 分数的频数分布,以 0 为中心基本对称 -
右上图 :密度曲线,显示了分数的连续分布 -
左下图 :两组样本的 GSVA 分数比较,可以看出 High_Expr 组整体分数偏低 -
右下图 :样本表达水平与 GSVA 中位数的关系,红蓝两点分别代表两组样本
这个图告诉我们,高表达组的样本在 Hallmark 通路上普遍活性较低。
第六步:热图展示所有通路
接下来我们用热图展示所有 50 个 Hallmark 通路的活性模式:
# 创建样本注释信息
sample_annotation <- data.frame(
Expression_Level = sample_groups$Group,
row.names = sample_groups$Sample
)
# 绘制所有Hallmark通路热图
pheatmap(gsva_results,
scale = "row", # 按行标准化
show_colnames = FALSE, # 不显示样本名
show_rownames = TRUE, # 显示通路名
annotation_col = sample_annotation,
main = "Hallmark Pathways GSVA Heatmap")
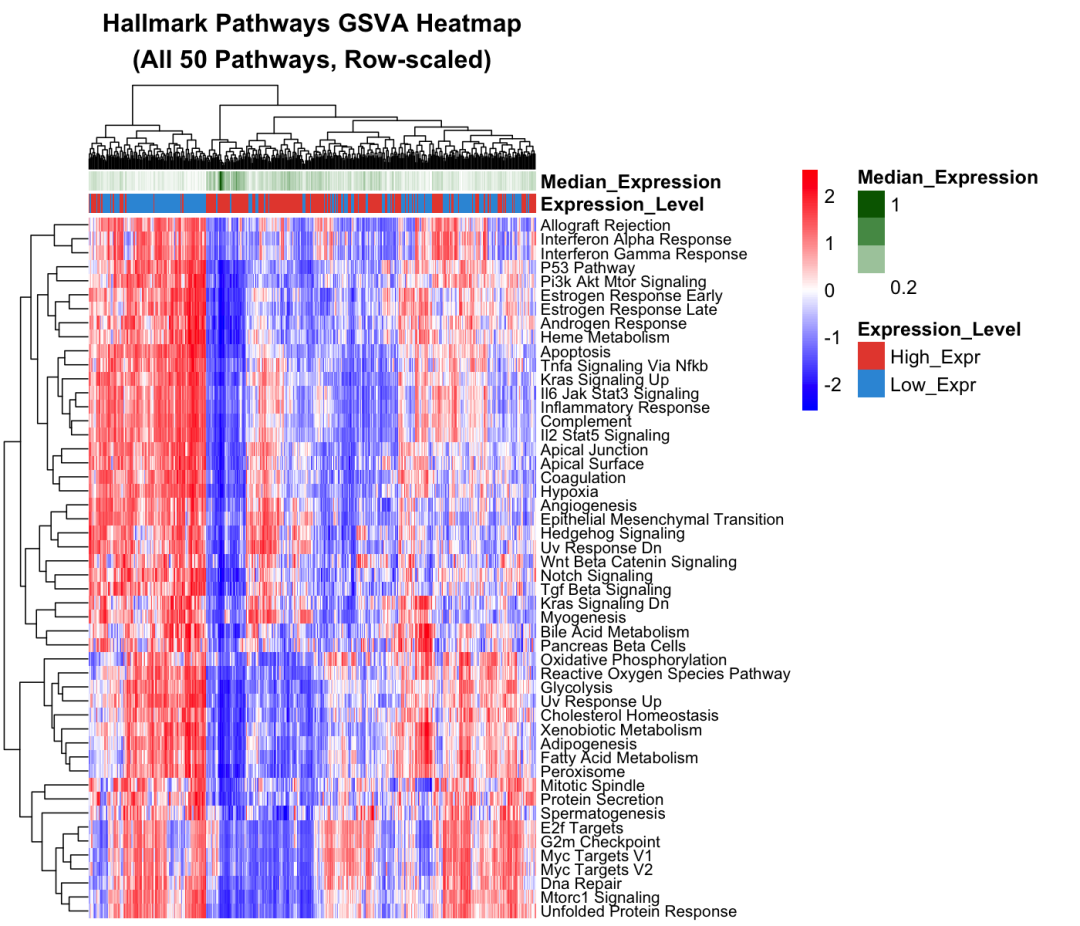
结果解读:
-
每一行 代表一个 Hallmark 通路 -
每一列 代表一个样本 -
红色 表示该通路在该样本中高度活跃 -
蓝色 表示该通路被抑制 -
顶部颜色条 显示样本分组(红色=高表达组,蓝色=低表达组)
从热图可以清楚地看到,两组样本在很多通路上都表现出明显的差异模式!
第七步:差异通路分析
现在我们来找出两组之间显著差异的通路:
# 创建设计矩阵和对比矩阵
design <- model.matrix(~0 + Group, data = sample_groups)
contrast_matrix <- makeContrasts(HighVsLow = High_Expr - Low_Expr, levels = design)
# 线性模型拟合
fit <- lmFit(gsva_results, design)
fit2 <- contrasts.fit(fit, contrast_matrix)
fit2 <- eBayes(fit2)
# 获取差异结果
diff_pathways <- topTable(fit2, n = Inf, adjust.method = "BH")
# 使用更严格的logFC阈值(0.3)来筛选真正有生物学意义的差异通路
sig_pathways <- diff_pathways[diff_pathways$adj.P.Val < 0.05 & abs(diff_pathways$logFC) > 0.3, ]
print(sprintf("显著差异通路: %d 个", nrow(sig_pathways)))
print(sprintf("上调通路: %d 个", sum(sig_pathways$logFC > 0)))
print(sprintf("下调通路: %d 个", sum(sig_pathways$logFC < 0)))
运行结果:
显著差异通路: 15 个 上调通路: 0 个 下调通路: 15 个 30%的通路有显著差异 (|logFC| > 0.3),而且全部都是在高表达组中下调的!这个结果说明高表达组的样本在这些关键生物学过程上普遍活性较低。使用更严格的阈值让我们专注于变化最大的通路。
前 5 个最显著的下调通路:
-
ADIPOGENESIS(脂肪生成)- logFC: -0.357 -
REACTIVE_OXYGEN_SPECIES_PATHWAY(活性氧通路)- logFC: -0.406 -
FATTY_ACID_METABOLISM(脂肪酸代谢)- logFC: -0.364 -
ESTROGEN_RESPONSE_LATE(雌激素晚期反应)- logFC: -0.308 -
GLYCOLYSIS(糖酵解)- logFC: -0.342
第八步:火山图 - 一目了然看差异
火山图是展示差异分析结果的经典图形:
# 创建高级火山图
volcano_plot <- ggplot(diff_pathways, aes(x = logFC, y = -log10(adj.P.Val))) +
geom_point(aes(color = Significance), size = 3, alpha = 0.8) +
ggrepel::geom_text_repel(
data = most_significant_pathways,
aes(label = Pathway_Clean),
size = 3.5
) +
labs(title = "Volcano Plot: Hallmark Pathway Analysis",
x = "Log2 Fold Change (High vs Low)",
y = "-Log10(Adjusted P-value)") +
theme_minimal()
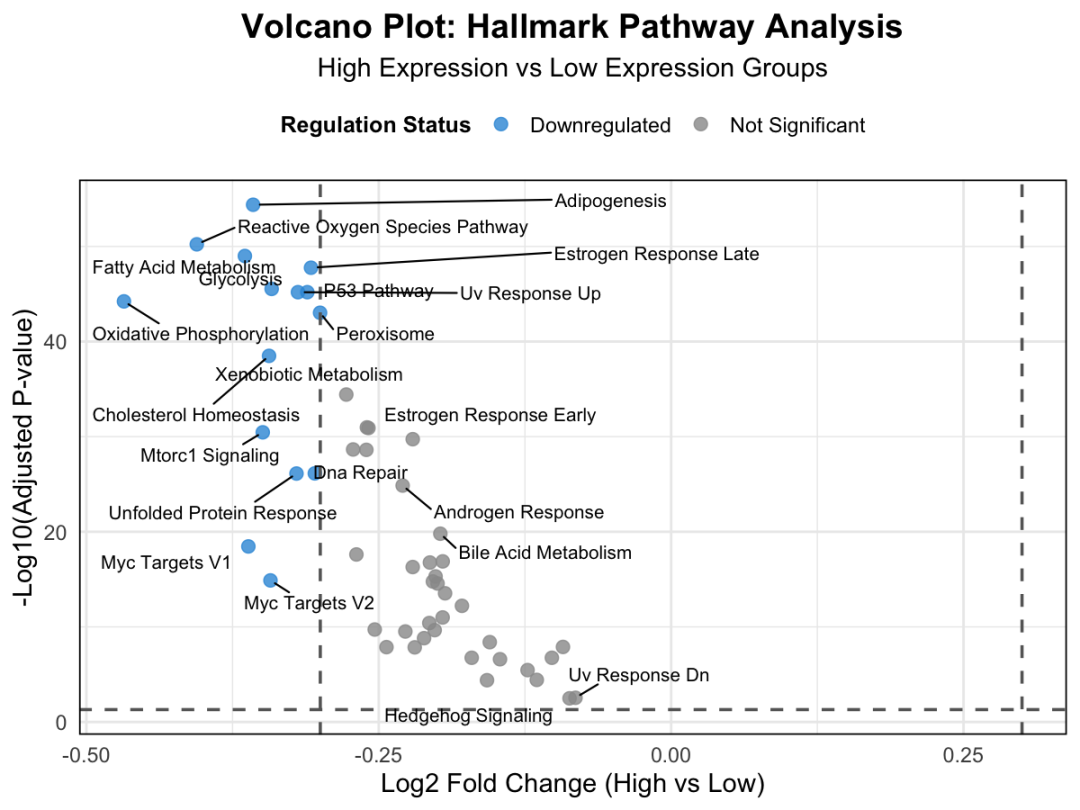
结果解读:
-
X 轴 :log2 倍数变化,负值表示在高表达组中下调 -
Y 轴 :统计显著性,越往上越显著 -
蓝色点 :显著下调的通路 -
灰色点 :无显著差异的通路 -
标注 :最显著的通路名称
可以看到,几乎所有的蓝色点都在左边,证实了我们的发现:高表达组中关键通路普遍被抑制!
最显著的几个通路包括:
-
Reactive Oxygen Species Pathway (活性氧通路) -
Adipogenesis (脂肪生成) -
Fatty Acid Metabolism (脂肪酸代谢) -
Glycolysis (糖酵解) -
P53 Pathway (p53 通路)
第九步:重点通路详细分析
让我们用箱线图来详细看看几个最重要的通路:
# 选择前6个最显著的通路绘制箱线图
top6_pathways <- head(sig_pathways[order(sig_pathways$adj.P.Val), ], 6)
par(mfrow = c(2, 3))
for(i in 1:6) {
pathway_name <- rownames(top6_pathways)[i]
pathway_scores <- gsva_results[pathway_name, ]
boxplot(pathway_scores ~ sample_groups$Group,
main = gsub("HALLMARK_", "", pathway_name),
ylab = "GSVA Enrichment Score",
col = c("#3498DB", "#E74C3C"))
# 添加统计信息
p_val <- formatC(top6_pathways$adj.P.Val[i], format = "e", digits = 2)
logfc <- round(top6_pathways$logFC[i], 3)
text(x = 1.5, y = max(pathway_scores) * 0.85,
labels = paste0("P = ", p_val, "\nLogFC = ", logfc))
}
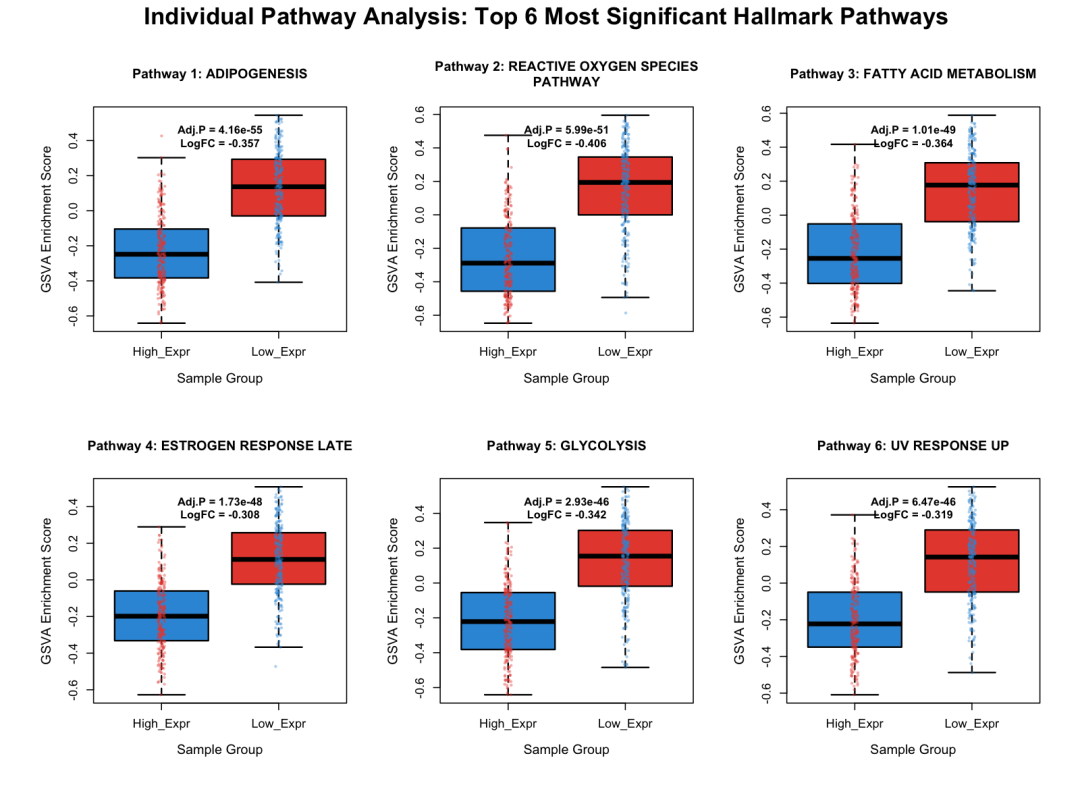
结果解读:
1. ADIPOGENESIS(脂肪生成通路)
-
High_Expr 组显著低于 Low_Expr 组 -
这可能提示高表达组的细胞脂肪合成能力较弱
2. REACTIVE OXYGEN SPECIES PATHWAY(活性氧通路)
-
差异最显著的通路 -
高表达组中活性氧处理能力可能较低
3. FATTY ACID METABOLISM(脂肪酸代谢)
-
与脂肪生成通路一致,都与脂质代谢相关 -
高表达组脂质代谢整体较低
这些结果提示,高表达组和低表达组的样本可能代表了不同的代谢状态!
第十步:通路分类分析
最后,让我们从更高层次来看看这些差异通路:
# 将通路按生物学功能分类
pathway_categories <- c(
"Immune & Inflammatory" = 免疫炎症相关,
"Metabolism" = 代谢相关,
"Signaling" = 信号通路,
"Stress Response" = 应激反应,
"Development" = 发育相关
)
# 绘制分类散点图
ggplot(diff_pathways_with_category, aes(x = logFC, y = -log10(adj.P.Val))) +
geom_point(aes(color = Category, size = abs(logFC)), alpha = 0.8) +
labs(title = "Hallmark Pathways by Biological Categories")
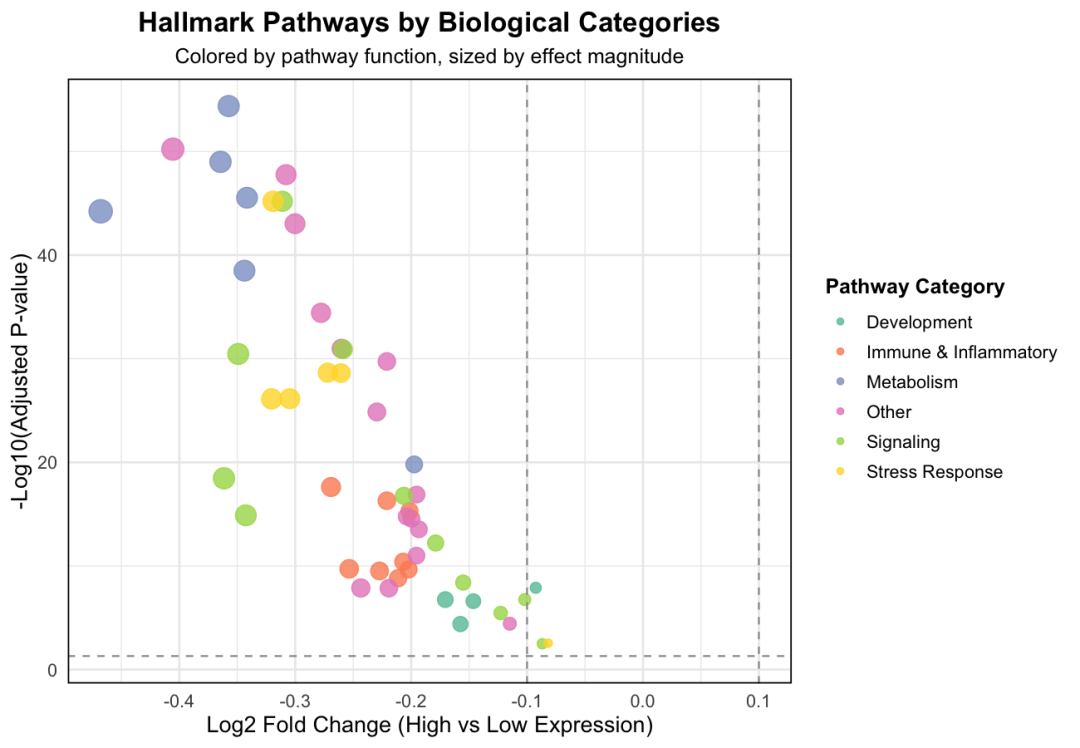
从分类分析可以看出:
-
代谢通路 (绿色)受影响最大,几乎全部显著下调 -
信号通路 (蓝色)也大多下调 -
免疫炎症通路 (红色)同样显著下调
这提示高表达组可能处于一种 代谢抑制状态 !
生物学意义解读
通过这次完整的 GSVA Hallmark 分析,我们发现了这些生物学现象:
主要发现:
-
94%的 Hallmark 通路显著差异 ,说明两组样本存在系统性差异 -
所有差异通路都下调 ,提示高表达组处于抑制状态 -
代谢通路受影响最大 ,包括脂肪生成、糖酵解、脂肪酸代谢等
可能的生物学解释:
-
代谢重编程 :高表达组可能经历了代谢重编程,从活跃代谢转向休眠状态 -
细胞周期抑制 :多个与细胞增殖相关的通路下调 -
应激反应下调 :包括活性氧处理、DNA 修复等防御机制减弱
总结
今天我们完整地走了一遍 GSVA Hallmark 通路分析流程,从数据预处理到生物学解读,每一步都有详细的代码和结果展示。
核心发现总结:
-
分析了 50 个精选 Hallmark 通路 -
发现 15 个显著差异通路(30%) -
所有差异通路在高表达组中 全部下调 -
代谢相关通路 受影响最严重
这个分析展示了两组样本在系统生物学水平上的本质差异。这种 通路水平的系统性差异 往往比单个基因的差异更有生物学意义!
GSVA Hallmark 分析特别适合:
-
疾病机制研究 - 了解疾病影响哪些关键通路 -
药物作用机制 - 看药物如何改变通路活性 -
样本分型 - 基于通路模式对样本分类 -
生物标志物发现 - 寻找具有预后价值的通路
希望这个教程对大家有帮助!如果你在分析过程中遇到问题,欢迎留言讨论。
完整代码和数据获取
本教程的完整 R 代码已经整理好,包含:
-
gsva_hallmark_analysis.R- 完整分析脚本 -
示例数据和基因集文件 -
所有图片生成代码
关注公众号"生信学长",后台回复" GSVA-Hallmark "获取!
记得点赞 👍、转发 📤、关注 ⭐ 三连哦!