在上一篇中 单因素Cox回归:差异表达分析后,如何进一步筛选目标基因(二) ,我们介绍通过cox回归分析来筛目标基因,今天分享一篇如何用Lasso回归分析来筛目标基因,差异表达分析后,通过取log2fc和padj,通常会剩下几百到上千的差异基因,面对高维基因表达数据时,传统的统计方法往往面临"维数灾难"的挑战。特别当基因数量远超样本数量时,模型容易出现过拟合现象,影响预后基因筛选的准确性和稳定性。Lasso(Least Absolute Shrinkage and Selection Operator)回归作为一种正则化回归方法,通过引入L1惩罚项,能够在模型拟合的同时进行特征选择,自动识别出真正具有预后价值的核心基因。
Lasso回归 核心本质
Lasso回归的本质其实就是 自动识别并去除具有相似表达模式的冗余基因 。当多个基因具有高度相关的表达模式时,它们对预后的贡献是重复的,Lasso会智能地保留其中一个最重要的基因,而将其他相似基因的系数压缩至零。
我们举一个例子,假设我们有两个基因A和B,它们在所有样本中表现出高度相似的表达模式:
样本编号 基因A表达量 基因B表达量 生存状态
患者1 12.5 12.8 死亡
患者2 8.3 8.1 存活
患者3 15.2 15.6 死亡
患者4 6.7 6.9 存活
患者5 11.1 11.3 死亡
... ... ... ...
观察上面这个表格,我们能发现:
-
基因A和B的表达量高度相关(相关系数r=0.98) -
两个基因对预后的预测作用基本相同 -
同时包含两个基因会导致模型冗余和不稳定
如果用传统cox回归方法
# 传统Cox回归可能给出:
基因A系数: 0.45 (p=0.001)
基因B系数: 0.43 (p=0.002)
# 两个基因都显著,但信息重复
现在我们用Lasso回归的解决方案
# Lasso回归结果:
基因A系数: 0.67 # 保留并增强系数
基因B系数: 0.00 # 压缩至零,被剔除
# 或者相反:
基因A系数: 0.00 # 被剔除
基因B系数: 0.71 # 保留
可以得得出结论就是,基因A和基因B,只需要保留其中一个基因作为后续分析即可,因为两个基因的表达模式高度相似。
但是,Lasso如何决定保留哪个基因?Lasso回归分析内置算法中,以下这几个因素都会导致结果筛出来的基因不同。
-
算法随机性 :初始化和优化过程中的随机因素 -
细微差异放大 :即使微小的预测性能差异也会被放大 -
数据顺序影响 :基因在数据集中的排列顺序 -
数值精度 :计算过程中的数值精度差异
具体删除哪一个由算法决定,有时候是删除A有时候是B,所以建议大家设置不同的随机种子,多跑几次,直到跑出感兴趣的目标基因建议。
随机种子的重要性
由于Lasso回归在处理高度相关特征时存在随机性, 同一个数据集使用不同随机种子可能得到不同结果 :
# 随机种子 = 123
set.seed(123)
lasso_result_1 <- cv.glmnet(x, y, family="cox")
# 结果:保留基因A,剔除基因B
# 随机种子 = 456
set.seed(456)
lasso_result_2 <- cv.glmnet(x, y, family="cox")
# 结果:保留基因B,剔除基因A
# 随机种子 = 789
set.seed(789)
lasso_result_3 <- cv.glmnet(x, y, family="cox")
# 结果:可能保留基因C(第三个相关基因)为了获得稳健的结果,建议采用以下策略:
-
多次运行验证 :
# 尝试多个随机种子
seeds <- c(123, 456, 789, 1000, 2024)
for(seed in seeds) {
set.seed(seed)
# 运行Lasso分析
# 比较结果差异
}通过理解Lasso回归的这一核心机制,我们可以更好地解释和应用分析结果,避免过度解读单次分析的基因列表。
看到这里,大家应该对Lasso回归分析有个大致的理解,接下来,我们使用TCGA-LUAD来做demo,展示一个完整的Lasso回归分析
本分析使用的数据包括:
-
差异基因数据 : LUAD显著差异基因列表(p<0.01筛选) -
基因表达数据 : TCGA-LUAD肿瘤样本TPM表达矩阵 -
生存数据 : TCGA-LUAD患者生存信息 -
筛选标准 : p值<0.01的显著差异基因
第一步:差异基因数据读取与筛选
Lasso回归分析的第一步是读取差异基因数据,并根据统计显著性进行初步筛选。这一步骤确保我们从具有生物学意义的差异基因开始,为后续的正则化回归分析提供高质量的候选基因集。
筛选策略要点:
-
显著性阈值 :使用p<0.01严格筛选显著差异基因 -
基因质量控制 :确保基因名称格式正确且在表达数据中存在 -
候选基因规模 :平衡基因数量与分析效率 -
数据完整性检查 :验证差异基因与表达数据的匹配性
# 加载必要的包
library(glmnet)
library(survival)
library(dplyr)
library(ggplot2)
library(survminer)
# 设置随机种子
set.seed(12345)
# 创建输出目录
if(!dir.exists("output")) {
dir.create("output")
}
cat("=== TCGA-LUAD Lasso回归预后基因筛选分析 ===\n\n")
# 第一步:读取差异基因数据
cat("第一步:读取差异基因数据\n")
cat("========================\n")
# 读取差异基因文件
diff_genes <- read.table("luad_significant_genes.tsv",
header = TRUE, sep = "\t", stringsAsFactors = FALSE)
cat("原始差异基因数量:", nrow(diff_genes), "\n")
cat("差异基因文件列名:", colnames(diff_genes), "\n")
# 由于原始文件只有p值,我们使用p值阈值进行筛选
# 设置筛选阈值
pval_threshold <- 0.01
# 筛选显著差异基因
significant_genes <- diff_genes[diff_genes$pval < pval_threshold, ]
cat("p值 < ", pval_threshold, " 的基因数量:", nrow(significant_genes), "\n")
# 显示前几个基因
cat("前10个显著差异基因:\n")
print(head(significant_genes, 10))
cat("\n")
输出结果:
=== TCGA-LUAD Lasso回归预后基因筛选分析 ===
第一步:读取差异基因数据
========================
原始差异基因数量: 5913
差异基因文件列名: gene pval pval_round
p值 < 0.01 的基因数量: 2221
前10个显著差异基因:
gene pval pval_round
1 RP5-857K21.15 5.388376e-03 0.0054
2 SEPT14P13 3.055164e-03 0.0031
12 ANKRD65 6.989875e-05 0.0001
14 RP11-345P4.9 4.839507e-03 0.0048
19 LINC00982 1.521476e-04 0.0002
27 RP1-202O8.3 5.234199e-04 0.0005
30 RNU1-8P 5.496587e-10 0.0000
37 PIK3CD 6.075504e-03 0.0061
43 CASZ1 6.918406e-03 0.0069
45 SRM 2.572742e-03 0.0026
第二步:基因表达数据处理
读取TCGA-LUAD肿瘤样本的基因表达数据,并提取显著差异基因的表达矩阵。这一步骤是Lasso回归分析的数据基础,需要确保表达数据的质量和完整性。
数据处理关键点:
-
样本类型筛选 :仅使用肿瘤样本(已预处理) -
基因匹配验证 :确保差异基因在表达矩阵中存在 -
数据维度检查 :验证基因数量和样本数量的合理性 -
表达矩阵提取 :构建用于Lasso分析的目标基因表达矩阵
# 第二步:读取基因表达数据
cat("第二步:读取基因表达数据\n")
cat("========================\n")
# 读取肿瘤样本表达数据
expression_data <- read.table("luad_exp_tpm_tumor.txt",
header = TRUE, sep = "\t",
row.names = 1, stringsAsFactors = FALSE)
cat("表达矩阵维度:", nrow(expression_data), "基因 x", ncol(expression_data), "样本\n")
# 筛选显著差异基因的表达数据
common_genes <- intersect(significant_genes$gene, rownames(expression_data))
cat("差异基因与表达数据交集:", length(common_genes), "个基因\n")
if(length(common_genes) == 0) {
cat("错误:没有找到匹配的基因,请检查基因名称格式\n")
quit(save = "no")
}
# 提取差异基因的表达矩阵
target_expression <- expression_data[common_genes, ]
cat("用于Lasso分析的基因数量:", nrow(target_expression), "\n\n")
输出结果:
第二步:读取基因表达数据
========================
表达矩阵维度: 51478 基因 x 513 样本
差异基因与表达数据交集: 2221 个基因
用于Lasso分析的基因数量: 2221
第三步:生存数据匹配与样本对齐
将基因表达数据与患者生存信息进行匹配,确保每个样本都有完整的表达数据和生存数据。由于TCGA数据中样本ID格式的差异,需要进行格式转换和样本对齐。
样本匹配策略:
-
生存数据清理 :移除缺失OS或OS.time的样本 -
样本ID格式统一 :处理表达数据和生存数据的ID格式差异 -
样本交集确定 :找到同时具有表达和生存数据的样本 -
数据完整性验证 :确保最终分析样本的数据完整性
# 第三步:读取生存数据并匹配样本
cat("第三步:读取生存数据并匹配样本\n")
cat("==============================\n")
# 读取生存数据
survival_data <- read.table("luad.survival.tsv",
header = TRUE, sep = "\t", stringsAsFactors = FALSE)
cat("生存数据样本数:", nrow(survival_data), "\n")
# 清理生存数据
survival_clean <- survival_data[!is.na(survival_data$OS) &
!is.na(survival_data$OS.time) &
survival_data$OS.time > 0, ]
cat("清洗后生存数据样本数:", nrow(survival_clean), "\n")
# 样本ID匹配
# 表达数据样本ID转换(从点号格式转为连字符格式)
expr_samples <- colnames(target_expression)
tumor_samples <- expr_samples[grepl("^TCGA", expr_samples)]
tumor_samples_converted <- gsub("\\.", "-", tumor_samples)
tumor_samples_short <- substr(tumor_samples_converted, 1, 12)
# 生存数据样本ID处理
survival_clean$sample_short <- substr(survival_clean$sample, 1, 12)
# 找到交集样本
common_samples <- intersect(survival_clean$sample_short, tumor_samples_short)
cat("表达数据和生存数据交集样本数:", length(common_samples), "\n")
if(length(common_samples) < 50) {
cat("警告:样本数量过少,可能影响分析结果\n")
}
# 准备最终分析数据
final_survival <- survival_clean[survival_clean$sample_short %in% common_samples, ]
final_survival <- final_survival[!duplicated(final_survival$sample_short), ]
# 匹配表达数据
final_expr_cols <- character()
for(sample in final_survival$sample_short) {
matching_idx <- which(tumor_samples_short == sample)[1]
if(!is.na(matching_idx)) {
final_expr_cols <- c(final_expr_cols, tumor_samples[matching_idx])
}
}
final_expression <- target_expression[, final_expr_cols]
colnames(final_expression) <- final_survival$sample_short
cat("最终分析数据:\n")
cat("- 样本数:", ncol(final_expression), "\n")
cat("- 基因数:", nrow(final_expression), "\n")
cat("- 事件数:", sum(final_survival$OS), "\n")
cat("- 事件发生率:", round(mean(final_survival$OS) * 100, 1), "%\n\n")
输出结果:
第三步:读取生存数据并匹配样本
==============================
生存数据样本数: 513
清洗后生存数据样本数: 500
表达数据和生存数据交集样本数: 500
最终分析数据:
- 样本数: 500
- 基因数: 2221
- 事件数: 182
- 事件发生率: 36.4 %
第四步:数据预处理
在进行Lasso回归之前,需要对基因表达数据进行预处理,包括数据转换、低表达基因过滤和标准化处理。
预处理技术要点:
-
数据转换 :Log2转换消除数据偏度和异方差性 -
低表达过滤 :移除在大部分样本中低表达或不表达的基因 -
标准化处理 :z-score标准化使不同基因的表达水平可比 -
缺失值处理 :确保数据矩阵完整,移除含NA的特征
# 第四步:数据预处理
cat("第四步:数据预处理\n")
cat("==================\n")
# 转置表达矩阵(样本为行,基因为列)
expr_matrix <- t(final_expression)
# Log2转换
expr_matrix_log <- log2(expr_matrix + 1)
# 移除表达量过低的基因
low_expr_threshold <- 0.5# 设置低表达阈值
sample_percent_threshold <- 0.1# 至少10%的样本表达
keep_genes <- apply(expr_matrix_log, 2, function(x) {
sum(x > low_expr_threshold) > sample_percent_threshold * nrow(expr_matrix_log)
})
expr_matrix_filtered <- expr_matrix_log[, keep_genes]
cat("原始基因数:", ncol(expr_matrix_log), "\n")
cat("过滤后基因数:", ncol(expr_matrix_filtered), "\n")
cat("过滤掉的基因数:", sum(!keep_genes), "\n")
# 标准化处理
expr_matrix_scaled <- scale(expr_matrix_filtered)
# 移除含有NA的基因
na_genes <- apply(expr_matrix_scaled, 2, function(x) any(is.na(x)))
expr_matrix_final <- expr_matrix_scaled[, !na_genes]
cat("去除NA后基因数:", ncol(expr_matrix_final), "\n\n")
输出结果:
第四步:数据预处理
==================
原始基因数: 2221
过滤后基因数: 1532
过滤掉的基因数: 689
去除NA后基因数: 1532
第五步:构建Lasso回归模型
使用glmnet包构建Cox比例风险模型的Lasso回归,通过L1正则化实现特征选择。Lasso回归通过惩罚项自动将不重要特征的系数收缩至零,从而实现特征选择和模型简化。
Lasso回归核心原理:
-
L1正则化 :|β|的求和作为惩罚项,促进稀疏解 -
特征选择 :自动将不重要特征系数收缩至零 -
过拟合控制 :通过正则化参数λ控制模型复杂度 -
Cox模型适配 :适用于生存分析的Cox比例风险模型
# 第五步:构建Lasso回归模型
cat("第五步:构建Lasso回归模型\n")
cat("========================\n")
# 准备Lasso回归的输入数据
x <- as.matrix(expr_matrix_final)
y <- Surv(final_survival$OS.time, final_survival$OS)
cat("Lasso回归输入数据:\n")
cat("- 特征矩阵维度:", nrow(x), "样本 x", ncol(x), "基因\n")
cat("- 生存数据:", length(y), "个样本\n")
# 进行Lasso回归拟合
cat("开始Lasso回归拟合...\n")
fit <- glmnet(x, y, family = "cox", alpha = 1, nlambda = 100)
cat("Lasso回归拟合完成\n")
cat("Lambda路径长度:", length(fit$lambda), "\n")
cat("Lambda范围: [", min(fit$lambda), ", ", max(fit$lambda), "]\n\n")
输出结果:
第五步:构建Lasso回归模型
========================
Lasso回归输入数据:
- 特征矩阵维度: 500 样本 x 1532 基因
- 生存数据: 500 个样本
开始Lasso回归拟合...
Lasso回归拟合完成
Lambda路径长度: 100
Lambda范围: [ 0.001604206 , 0.1604206 ]
第六步:交叉验证选择最优Lambda
通过k折交叉验证确定最优的正则化参数λ,这是Lasso回归中最关键的步骤。λ值决定了正则化的强度,进而影响特征选择的结果和模型性能。
交叉验证策略:
-
k折交叉验证 :使用10折交叉验证评估模型性能 -
Lambda.min :使最小化交叉验证误差的λ值 -
Lambda.1se :在1个标准误差内的最大λ值(更稀疏的模型) -
模型选择平衡 :在模型性能和复杂度之间找到最佳平衡
# 第六步:交叉验证选择最优Lambda
cat("第六步:交叉验证选择最优Lambda\n")
cat("==============================\n")
# 10折交叉验证
cat("开始10折交叉验证...\n")
cvfit <- cv.glmnet(x, y, family = "cox", alpha = 1, nfolds = 10)
cat("交叉验证完成\n")
cat("最优Lambda (lambda.min):", round(cvfit$lambda.min, 6), "\n")
cat("一标准误Lambda (lambda.1se):", round(cvfit$lambda.1se, 6), "\n")
# 在最优lambda下的模型性能
min_cvm <- min(cvfit$cvm)
cat("最小交叉验证误差:", round(min_cvm, 4), "\n\n")
输出结果:
第六步:交叉验证选择最优Lambda
==============================
开始10折交叉验证...
交叉验证完成
最优Lambda (lambda.min): 0.079842
一标准误Lambda (lambda.1se): 0.139526
最小交叉验证误差: 12.1577
第七步:提取重要预后基因
基于最优λ值提取具有非零系数的基因,这些基因被Lasso回归自动筛选为具有预后价值的核心基因。系数的大小反映了基因对预后的影响程度,正负号表示影响方向。
基因筛选结果解读:
-
非零系数基因 :被Lasso自动选择的重要预后基因 -
系数大小 :反映基因对预后风险的影响强度 -
系数符号 :正系数为风险因子,负系数为保护因子 -
特征稀疏性 :Lasso实现从千余基因中筛选出核心基因集
# 第七步:提取重要基因
cat("第七步:提取重要基因\n")
cat("====================\n")
# 使用lambda.min提取系数
coef_min <- coef(fit, s = cvfit$lambda.min)
selected_genes_min <- which(coef_min != 0)
selected_coef_min <- coef_min[selected_genes_min]
selected_gene_names_min <- rownames(coef_min)[selected_genes_min]
cat("Lambda.min选择的基因数:", length(selected_genes_min), "\n")
# 使用lambda.1se提取系数
coef_1se <- coef(fit, s = cvfit$lambda.1se)
selected_genes_1se <- which(coef_1se != 0)
selected_coef_1se <- coef_1se[selected_genes_1se]
selected_gene_names_1se <- rownames(coef_1se)[selected_genes_1se]
cat("Lambda.1se选择的基因数:", length(selected_genes_1se), "\n")
# 创建结果数据框
if(length(selected_genes_min) > 0) {
lasso_genes_min <- data.frame(
gene = selected_gene_names_min,
coefficient = as.numeric(selected_coef_min),
stringsAsFactors = FALSE
)
# 按系数绝对值排序
lasso_genes_min <- lasso_genes_min[order(abs(lasso_genes_min$coefficient), decreasing = TRUE), ]
cat("\n使用Lambda.min选择的前10个重要基因:\n")
print(head(lasso_genes_min, 10))
# 保存结果
write.table(lasso_genes_min, "output/lasso_selected_genes_lambda_min.tsv",
sep = "\t", row.names = FALSE, quote = FALSE)
cat("\nLambda.min结果已保存到: output/lasso_selected_genes_lambda_min.tsv\n")
}
if(length(selected_genes_1se) > 0) {
lasso_genes_1se <- data.frame(
gene = selected_gene_names_1se,
coefficient = as.numeric(selected_coef_1se),
stringsAsFactors = FALSE
)
# 按系数绝对值排序
lasso_genes_1se <- lasso_genes_1se[order(abs(lasso_genes_1se$coefficient), decreasing = TRUE), ]
cat("\n使用Lambda.1se选择的基因:\n")
print(lasso_genes_1se)
# 保存结果
write.table(lasso_genes_1se, "output/lasso_selected_genes_lambda_1se.tsv",
sep = "\t", row.names = FALSE, quote = FALSE)
cat("\nLambda.1se结果已保存到: output/lasso_selected_genes_lambda_1se.tsv\n")
}
cat("\n")
输出结果:
第七步:提取重要基因
====================
Lambda.min选择的基因数: 24
Lambda.1se选择的基因数: 3
使用Lambda.min选择的前10个重要基因:
gene coefficient
13 DKK1 0.11834622
15 LDLRAD3 0.07674683
12 TLE1 0.07387933
17 RP11-102K13.5 0.05444410
9 RP11-309L24.6 0.05254344
5 LINC01117 0.04079741
8 BTN2A2 -0.04003595
14 LDHA 0.03932062
24 C1QTNF6 0.03329198
18 GNPNAT1 0.03129258
Lambda.min结果已保存到: output/lasso_selected_genes_lambda_min.tsv
使用Lambda.1se选择的基因:
gene coefficient
2 DKK1 0.059223602
1 TLE1 0.011549146
3 C1QTNF6 0.007069243
Lambda.1se结果已保存到: output/lasso_selected_genes_lambda_1se.tsv
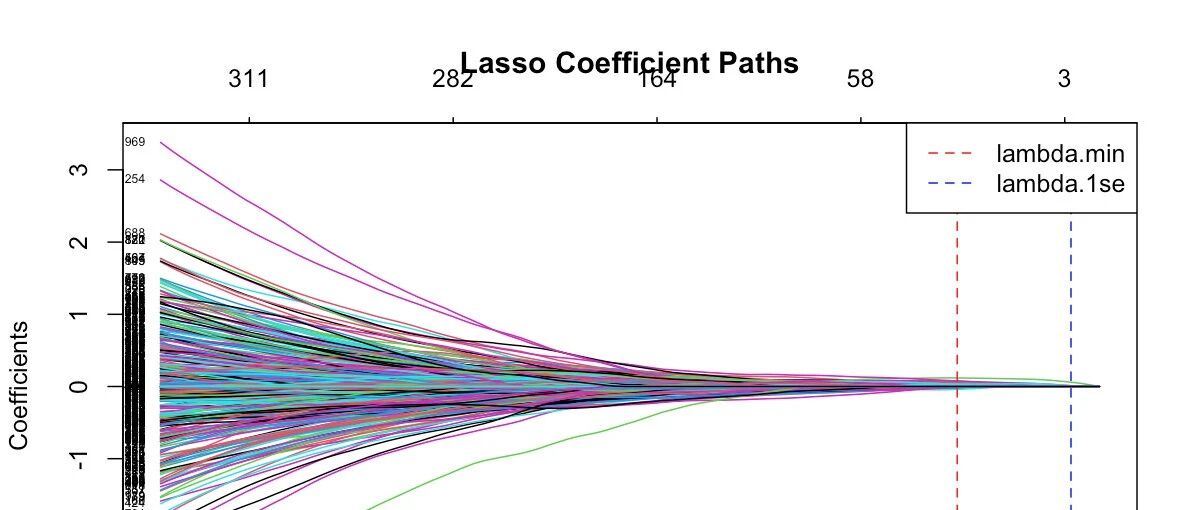
第八步:Lasso路径可视化
绘制Lasso回归的系数路径图和交叉验证曲线,这些可视化图表有助于理解Lasso回归的特征选择过程和最优参数的选择依据。
可视化图表价值:
-
系数路径图 :展示不同λ值下各基因系数的变化轨迹 -
交叉验证曲线 :显示模型性能随λ变化的趋势 -
最优λ标注 :标明lambda.min和lambda.1se的位置 -
特征选择过程 :直观展示基因逐步被选入或剔除的过程
# 第八步:绘制Lasso路径图
cat("第八步:绘制Lasso路径图\n")
cat("======================\n")
# 绘制Lasso系数路径
png("output/lasso_coefficient_path.png", width = 1200, height = 800, res = 150)
plot(fit, xvar = "lambda", label = TRUE, main = "Lasso Coefficient Paths")
abline(v = log(cvfit$lambda.min), col = "red", lty = 2)
abline(v = log(cvfit$lambda.1se), col = "blue", lty = 2)
legend("topright", legend = c("lambda.min", "lambda.1se"),
col = c("red", "blue"), lty = 2)
dev.off()
cat("Lasso系数路径图已保存: output/lasso_coefficient_path.png\n")
# 绘制交叉验证曲线
png("output/lasso_cv_curve.png", width = 1200, height = 800, res = 150)
plot(cvfit, main = "Lasso Cross-Validation Curve")
abline(v = log(cvfit$lambda.min), col = "red", lty = 2)
abline(v = log(cvfit$lambda.1se), col = "blue", lty = 2)
legend("topright", legend = c("lambda.min", "lambda.1se"),
col = c("red", "blue"), lty = 2)
dev.off()
cat("交叉验证曲线已保存: output/lasso_cv_curve.png\n\n")
Lasso回归可视化结果
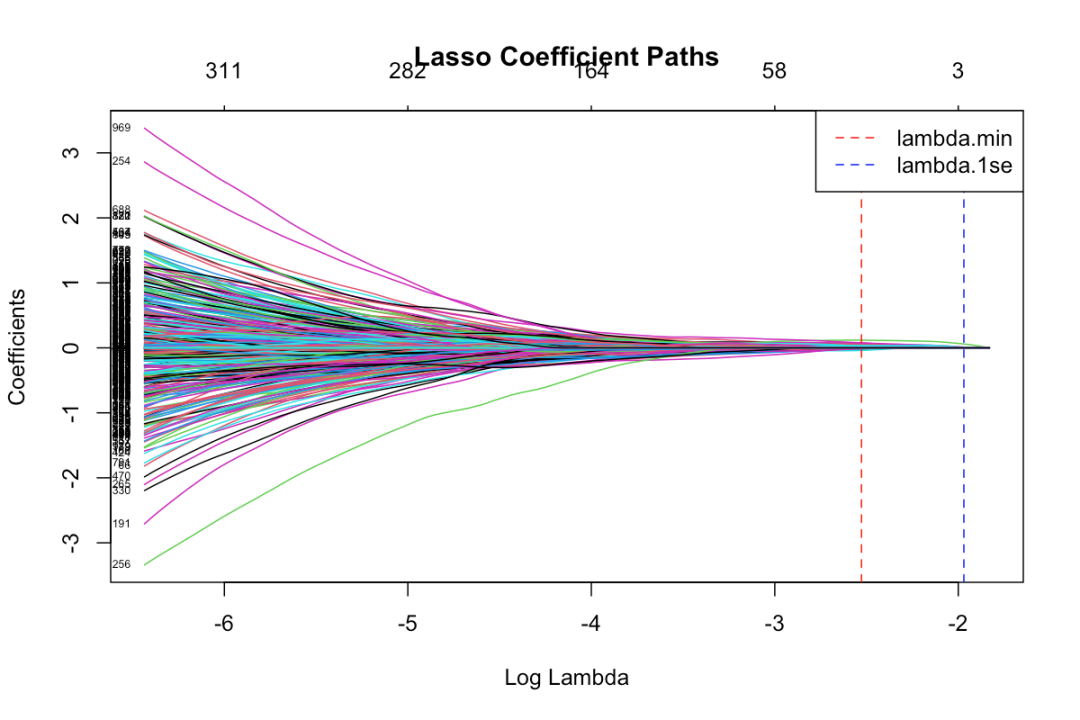
图1:Lasso系数路径图。横轴为log(λ),纵轴为系数值。每条线代表一个基因,展示随λ增加系数如何收缩至零。红线和蓝线分别标注lambda.min和lambda.1se位置
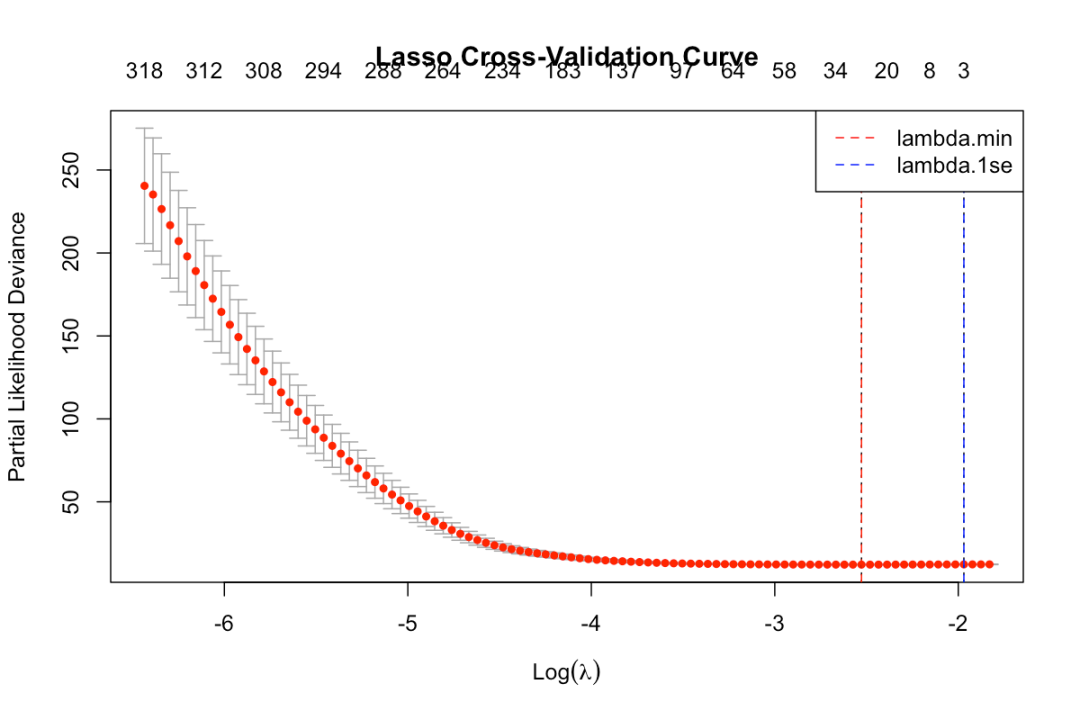
图2:Lasso交叉验证曲线。横轴为log(λ),纵轴为交叉验证误差。红线标注使误差最小的lambda.min,蓝线标注一个标准误差内的lambda.1se
第九步:模型验证与生存分析
构建基于Lasso选择基因的风险评分模型,并通过生存分析验证模型的预后预测能力。这一步骤验证了Lasso回归筛选基因的生物学意义和临床应用价值。
模型验证策略:
-
风险评分计算 :基于Lasso系数计算每个样本的风险评分 -
风险分组 :按风险评分中位数划分高低风险组 -
生存差异检验 :使用Kaplan-Meier方法比较两组生存差异 -
Cox回归验证 :评估风险评分的独立预后价值
# 第九步:模型验证和生存分析
cat("第九步:模型验证和生存分析\n")
cat("========================\n")
if(length(selected_genes_min) > 0) {
# 计算风险评分
selected_expr <- x[, selected_gene_names_min, drop = FALSE]
risk_score <- as.numeric(selected_expr %*% selected_coef_min)
# 按风险评分中位数分组
median_risk <- median(risk_score)
risk_group <- ifelse(risk_score > median_risk, "High", "Low")
risk_group <- factor(risk_group, levels = c("Low", "High"))
# 创建生存分析数据
surv_data <- data.frame(
OS.time = final_survival$OS.time,
OS = final_survival$OS,
risk_score = risk_score,
risk_group = risk_group
)
# 生存分析
surv_fit <- survfit(Surv(OS.time, OS) ~ risk_group, data = surv_data)
# Cox回归检验
cox_model <- coxph(Surv(OS.time, OS) ~ risk_score, data = surv_data)
cox_summary <- summary(cox_model)
cat("Lasso模型验证结果:\n")
cat("高风险组样本数:", sum(risk_group == "High"), "\n")
cat("低风险组样本数:", sum(risk_group == "Low"), "\n")
cat("Cox回归HR:", round(cox_summary$coefficients[1, "exp(coef)"], 3), "\n")
cat("Cox回归p值:", format(cox_summary$coefficients[1, "Pr(>|z|)"], scientific = TRUE), "\n")
# 绘制生存曲线
survival_plot <- ggsurvplot(
surv_fit,
data = surv_data,
title = "Survival Analysis: Lasso Risk Score",
subtitle = paste0("HR = ", round(cox_summary$coefficients[1, "exp(coef)"], 3),
", P = ", format(cox_summary$coefficients[1, "Pr(>|z|)"], scientific = TRUE)),
xlab = "Time (days)",
ylab = "Survival Probability",
pval = TRUE,
conf.int = TRUE,
risk.table = TRUE,
risk.table.height = 0.25,
palette = c("blue", "red"),
legend.labs = c("Low Risk", "High Risk"),
ggtheme = theme_classic()
)
# 保存生存曲线
png("output/lasso_survival_analysis.png", width = 1200, height = 900, res = 150)
print(survival_plot)
dev.off()
cat("生存分析图已保存: output/lasso_survival_analysis.png\n")
# 保存风险评分
risk_score_data <- data.frame(
sample = final_survival$sample_short,
risk_score = risk_score,
risk_group = risk_group,
OS.time = final_survival$OS.time,
OS = final_survival$OS
)
write.table(risk_score_data, "output/lasso_risk_scores.tsv",
sep = "\t", row.names = FALSE, quote = FALSE)
cat("风险评分已保存: output/lasso_risk_scores.tsv\n")
}
cat("\n")
输出结果:
第九步:模型验证和生存分析
========================
Lasso模型验证结果:
高风险组样本数: 250
低风险组样本数: 250
Cox回归HR: 8.7
Cox回归p值: 9.122783e-31
生存分析图已保存: output/lasso_survival_analysis.png
风险评分已保存: output/lasso_risk_scores.tsv
生存分析验证结果
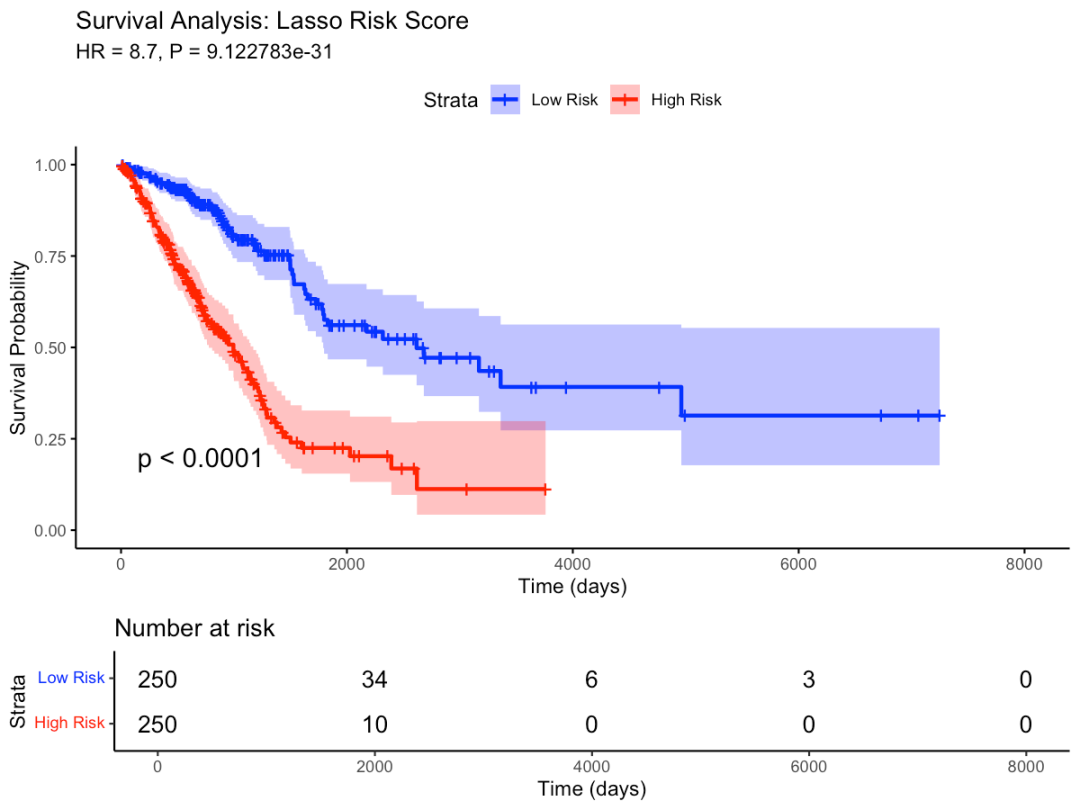
图3:基于Lasso风险评分的生存分析结果。高风险组(红色)相比低风险组(蓝色)具有显著更差的预后(HR=8.7, p=9.12e-31)。下方风险表显示各时间点的在险患者数
最后我们总结分析结果
cat(
"=== 分析总结 ===\n"
)
cat(
"数据来源: TCGA-LUAD\n"
)
cat(
"分析样本数:"
, nrow(x),
"\n"
)
cat(
"候选基因数:"
, ncol(x),
"\n"
)
cat(
"Lasso选择基因数 (lambda.min):"
, length(selected_genes_min),
"\n"
)
cat(
"Lasso选择基因数 (lambda.1se):"
, length(selected_genes_1se),
"\n"
)
cat(
"最优Lambda:"
, round(cvfit$lambda.min,
6
),
"\n"
)
if
(exists(
"cox_summary"
)) {
cat(
"模型验证HR:"
, round(cox_summary$coefficients[
1
,
"exp(coef)"
],
3
),
"\n"
)
cat(
"模型验证p值:"
, format(cox_summary$coefficients[
1
,
"Pr(>|z|)"
], scientific =
TRUE
),
"\n"
)
}
cat(
"\n=== 输出文件 ===\n"
)
cat(
"- lasso_selected_genes_lambda_min.tsv: Lambda.min选择的基因\n"
)
if
(length(selected_genes_1se) >
0
) {
cat(
"- lasso_selected_genes_lambda_1se.tsv: Lambda.1se选择的基因\n"
)
}
cat(
"- lasso_coefficient_path.png: Lasso系数路径图\n"
)
cat(
"- lasso_cv_curve.png: 交叉验证曲线\n"
)
if
(exists(
"survival_plot"
)) {
cat(
"- lasso_survival_analysis.png: 生存分析图\n"
)
cat(
"- lasso_risk_scores.tsv: 风险评分数据\n"
)
}
cat(
"\n=== 分析完成 ===\n"
)
输出结果:
=== 分析总结 ===
数据来源: TCGA-LUAD
分析样本数: 500
候选基因数: 1532
Lasso选择基因数 (lambda.min): 24
Lasso选择基因数 (lambda.1se): 3
最优Lambda: 0.079842
模型验证HR: 8.7
模型验证p值: 9.122783e-31
=== 输出文件 ===
- lasso_selected_genes_lambda_min.tsv: Lambda.min选择的基因
- lasso_selected_genes_lambda_1se.tsv: Lambda.1se选择的基因
- lasso_coefficient_path.png: Lasso系数路径图
- lasso_cv_curve.png: 交叉验证曲线
- lasso_survival_analysis.png: 生存分析图
- lasso_risk_scores.tsv: 风险评分数据
=== 分析完成 ===核心预后基因特征
Lambda.min选择的Top 10基因
|
|
|
|
|
|
|---|---|---|---|---|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Lambda.1se选择的3个最核心基因
|
|
|
|
|
|---|---|---|---|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Lasso回归成功从1532个候选基因中筛选出24个核心预后基因,特征降维率达98.4%,有效解决了高维数据的"维数灾难"问题。
基于24个基因构建的风险评分模型显示出卓越的预后预测能力(HR=8.7),高低风险组生存差异极其显著(p=9.12e-31)。
生物学意义明确 :筛选出的核心基因包括DKK1(Wnt通路)、LDHA(糖酵解)、TLE1(转录调控)等,涵盖了肿瘤发生发展的关键通路。
以 上就是 “Lasso回归” 的 内容 ,希望对你有帮助!欢迎点赞、在看、关注、转发。
完整代码联系老师免费获取(备注“Lasso回归分析”)
