今天来聊一下肿瘤微环境分析,我们在做bulk-RNA seq单细胞肿瘤分析时,经常需要做肿瘤微环境分析,来评估不同样本间免基质细胞、免疫细胞和肿瘤细胞的含量,进而可以根据不同细胞的含量,来判断不同分组样本间是否具有差异。
肿瘤微环境是什么
肿瘤微环境由多种细胞和非细胞成分组成,主要包括:
-
基质细胞 :如成纤维细胞,参与形成肿瘤的支持结构。 -
免疫细胞 :如T细胞、巨噬细胞,负责免疫监视和抗肿瘤反应。 -
血管系统 :为肿瘤提供营养和氧气,支持其生长。 -
细胞外基质 :提供结构支持,并参与细胞信号传导。
这些成分共同构成了肿瘤的生存环境,影响其生物学行为和治疗反应。
为什么要进行肿瘤微环境分析
肿瘤微环境可以评估不同样本间免基质细胞、免疫细胞和肿瘤细胞的含量,为后续做免疫细胞在不同组间的差异分析做数据准备。
下面我们讲一下R语言
estimate
包做肿瘤微环境分析的原理,本质就是一个数学模型来预测一块肿瘤组中各类细胞的含量。
estimate
包的打分原理基于特定基因集的表达水平,通过计算基质细胞、免疫细胞和肿瘤细胞相关基因的表达特征,分别生成基质评分(Stromal Score)、免疫评分(Immune Score)和肿瘤评分(Tumor Purity)。这些评分反映了肿瘤微环境中不同成分的相对比例和活跃程度,其中基质评分和免疫评分分别代表基质细胞和免疫细胞的浸润程度,而肿瘤评分则与肿瘤细胞的纯度呈负相关。
讲完原理,我们看下代码实现部分
如何使用R包
estimate
进行肿瘤微环境分析
estimate
是一个常用的R包,用于计算肿瘤微环境中基质细胞、免疫细胞和肿瘤细胞的评分。以下是具体步骤:
1.安装和加载R包
在R中安装并加载
estimate
包:
library(ggplot2)
library(limma)
library(estimate)
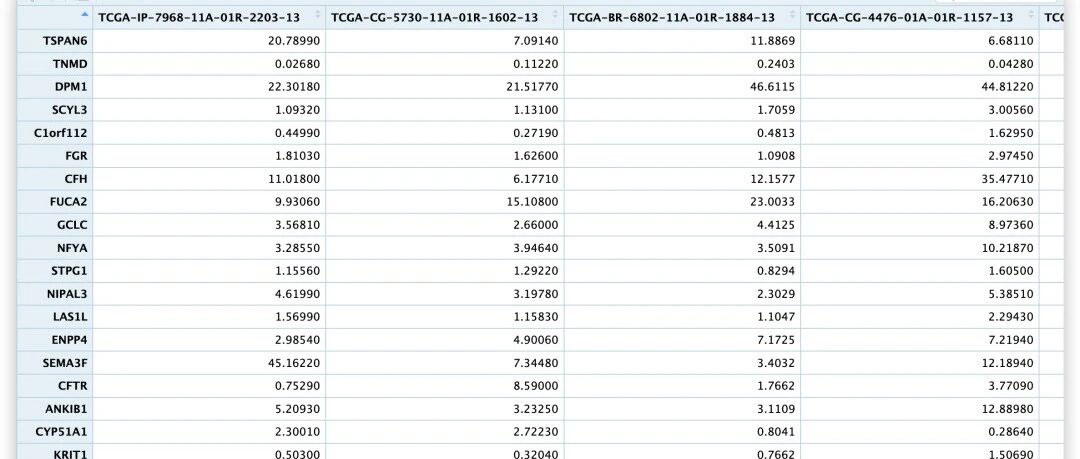
2.读取 基因表达矩阵,行代表基因,列代表样本。数据可以是RNA-seq或芯片数据
# 读取文件并整理输入文件
expression_data <- read.table("expression_matrix.txt", header = TRUE, sep = "\t", check.names = FALSE, row.names = 1) # 读取表达数据文件
expression_data <- as.matrix(expression_data) # 转换为矩阵
3.删除正常样品,只保留肿瘤样本
# 删除正常样品
sample_group <- sapply(strsplit(colnames(expression_data), "\\-"), "[", 4) # 提取组别信息
sample_group <- sapply(strsplit(sample_group, ""), "[", 1) # 提取组别的第一个字符
sample_group <- gsub("2", "1", sample_group) # 将2替换为1
expression_data <- expression_data[, sample_group == "0"] # 只保留肿瘤样品
# 输出整理好的数据
tumor_expression_matrix <- rbind(ID = colnames(expression_data), expression_data)
write.table(tumor_expression_matrix, file = "tumor_expression_matrix.tsv", sep = "\t", quote = FALSE, col.names = FALSE)
4.运行
estimate
函数
使用
estimateScore
函数计算评分:
filterCommonGenes(input.f = "tumor_expression_matrix.tsv", output.f = "common_genes.gct", id = "GeneSymbol") # 过滤常见基因
estimateScore(input.ds = "common_genes.gct", output.ds = "estimate_score.gct") # 计算估计分数
estimate_scores <- read.table("estimate_score.gct", skip = 2, header = TRUE) # 读取分数文件
该函数会返回一个包含基质细胞、免疫细胞和肿瘤细胞评分的矩阵。
我们打印
estimate_scores
变量,就是评分的结果
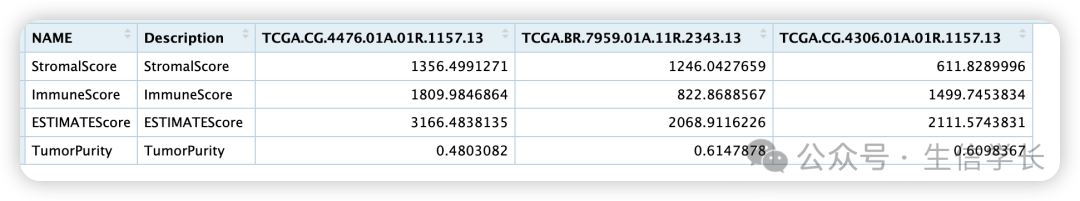
可以看到,1到4行分别是基质细胞评分、免疫细胞评分、综合评分、肿瘤纯度。我们将
estimate_scores
矩阵转置一下,输出一个行名为样本名称,列名为基质细胞评分、免疫细胞评分、综合评分、肿瘤纯度的矩阵
5.转置
estimate_scores
# 输出每个样品的打分
rownames(estimate_scores) <- estimate_scores[, 1]
transposed_scores <- t(estimate_scores[, 3:ncol(estimate_scores)])
rownames(transposed_scores) <- gsub("\\.", "\\-", rownames(transposed_scores))
# 保存到文件
final_output <- rbind(ID = colnames(transposed_scores), transposed_scores)
write.table(final_output, file = "tme_scores.tsv", sep = "\t", quote = FALSE, col.names = FALSE) # 保存为文件
# 打印转置后的表达矩阵
transposed_scores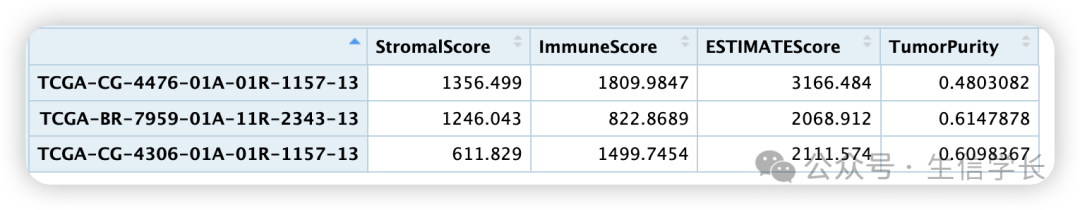
可以看到行名为不同样本的名称,1到4列分别为样本的基质细胞评分、免疫细胞评分、综合评分、肿瘤纯度。其中第3列
ESTIMATEScore
为综合评分,是前两列之和,分数越到表示肿瘤的纯度越低;第4列就是肿瘤的纯度。
这些指标评分反映了肿瘤微环境中不同成分的活跃程度:
-
基质细胞评分高 :提示肿瘤微环境中基质细胞活跃,可能与肿瘤转移相关。 -
免疫细胞评分高 :表明免疫细胞浸润较多,可能对免疫治疗有较好反应。 -
肿瘤细胞评分高 :反映肿瘤细胞占比高,可能与肿瘤恶性程度相关。
下一篇,我们讲一下如何做肿瘤微环境差异分析和小提琴图可视化
代码&文件联系作者,备注 “肿瘤微环境”

写作不易,欢迎关注