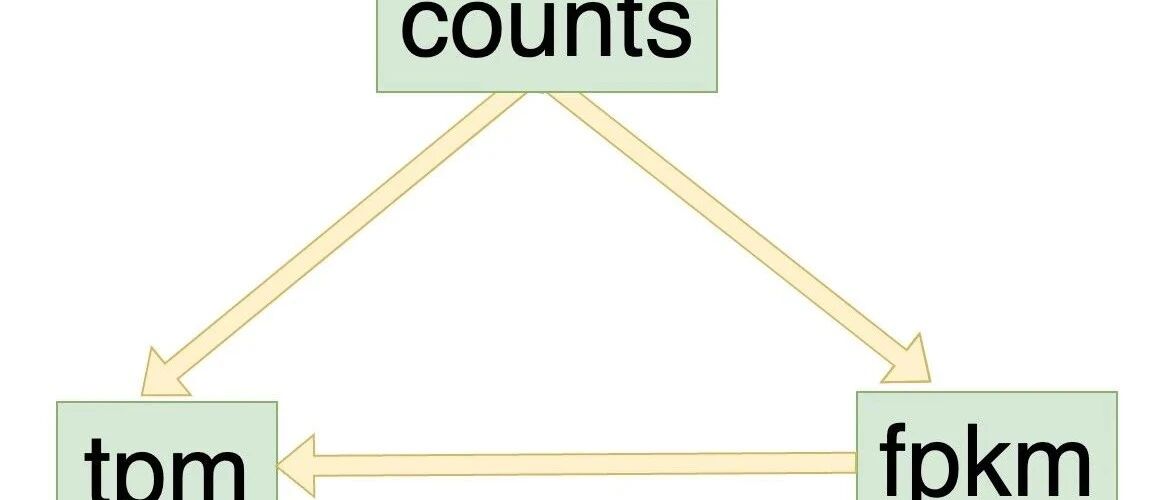
在 RNA 测序数据分析中,
counts
、
FPKM
、
TPM
是我们经常听到的概念。它们有什么区别?为什么我们不仅要用
counts
,还要用到
FPKM
和
TPM
呢?今天我们就来侃侃这些表达量的计算和区别。
Counts 值
首先来说说
counts
。在 RNA 测序中,
counts
是最原始的表达数据。我们把测序后比对到基因上的 reads 数量直接统计出来,这些数值就叫做 counts。简单来说,
counts
代表了每个基因在每个样本中的原始测序读数。
counts
值越高,说明这个基因在样本中表达越活跃。
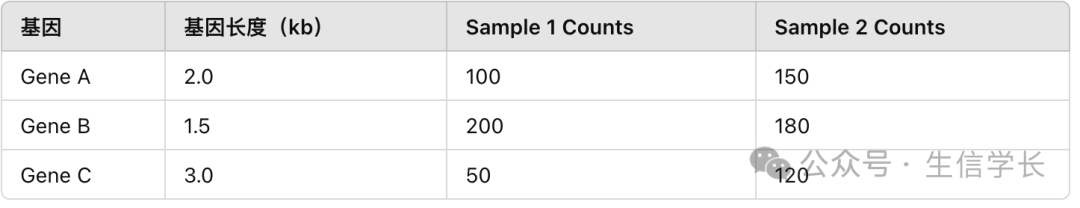
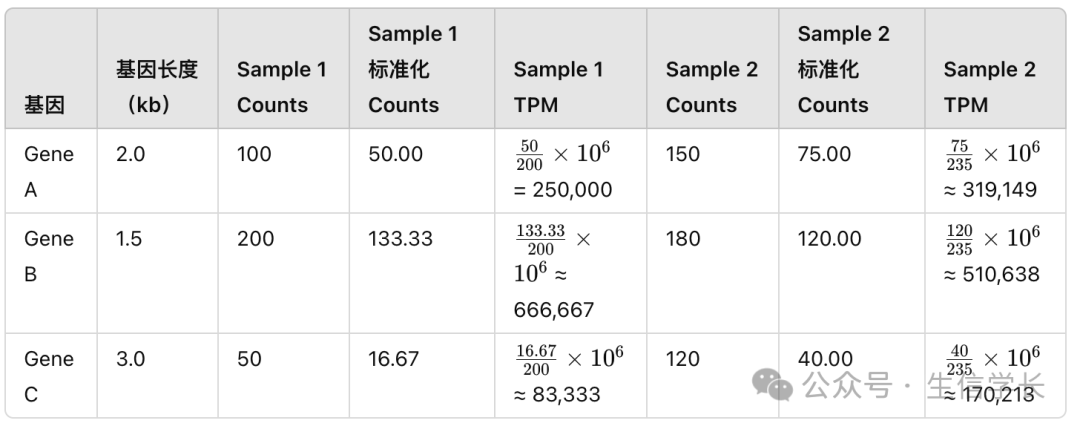
我们看一下下面三个基因(Gene A、Gene B、Gene C)和两个样本(Sample 1、Sample 2)的计数矩阵,
counts
表格如下:
-
Sample 1 Counts 和 Sample 2 Counts 表示的是原始的 reads 计数值。 -
基因长度表示的是每个基因的长度(以千碱基为单位,kb)。
有了Counts为什么还需要 FPKM 和 TPM?
counts
虽然是最原始的数值,但它的缺陷也很明显——受
基因长度
和
测序深度
的影响。比如,一个基因很长,那么它捕获到的 reads 数可能会更多,这样的
counts
值会偏高;而另一个基因虽然短但表达很强,反而可能因为长度原因捕获到的 reads 数比较少。这就让
counts
值不能直接反映基因的实际表达水平。
另外,测序深度也会影响
counts
值。不同样本的测序深度可能不一样,测得多的样本总 counts 值更高,看起来表达也会“虚高”。为了消除这些影响,才提出了
FPKM
和
TPM
这些标准化的方法,让我们能够更公平地比较基因的表达水平。
FPKM 的计算方式
FPKM(Fragments Per Kilobase of transcript per Million mapped reads)是一种校正方法,它的意思是“每千碱基的转录本在每百万 reads 中的片段数”。
FPKM 的计算公式如下:
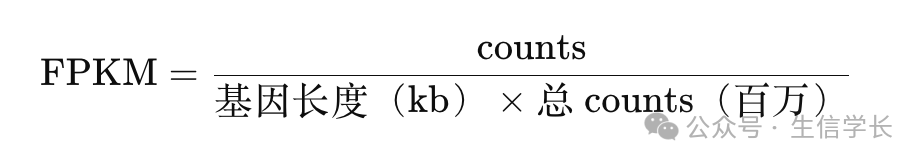
通过这个公式,FPKM 值先考虑了基因的长度,把 counts 除以基因的长度(以千碱基为单位),这样能让不同长度的基因具有可比性。接着,FPKM 值又除以样本的总 counts(以百万为单位),来控制测序深度的影响。因此,FPKM 能在一定程度上消除基因长度和测序深度的影响,适合用于同一个样本内的基因表达比较。
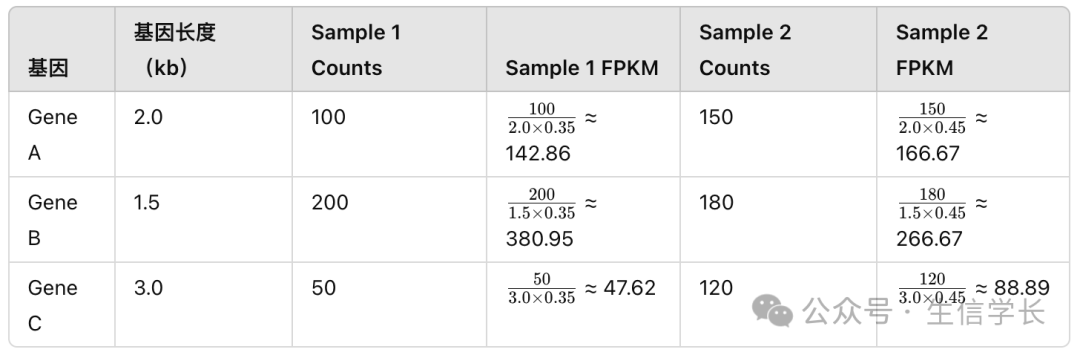
Sample 1 的总 counts 为
100 + 200 + 50 = 350
,Sample 2 的总 counts 为
150 + 180 + 120 = 450
我们可以计算出每个基因在两个样本中的 FPKM 值,结果如下:
为什么还需要 TPM
虽然 FPKM 校正了基因长度和测序深度的影响,但它并不适合用于不同样本间的比较。比如,两个样本各自的 FPKM 值并不完全反映真实的相对表达水平。因此,为了更精确地比较不同样本之间的基因表达,我们需要用到 TPM(Transcripts Per Million)。
TPM 其实和 FPKM 的思路类似,但计算方式稍有不同。它首先对每个基因做长度归一化,然后再把所有基因的标准化值相加,使总数为一百万。这样得到的 TPM 可以更好地在不同样本之间进行比较。
TPM 的计算方式
TPM(Transcripts Per Million) 是指“每百万个转录本中的某基因的转录本数”。它的计算方式分为两步:
-
基因长度归一化 :先将每个基因的 counts除以基因长度(以千碱基为单位),得到一个标准化的 counts 值。 -
总标准化值归一化 :将所有基因的标准化值相加,得到总标准化值。然后将每个基因的标准化值除以这个总值,并乘以一百万,得到 TPM 值。
公式如下:

这样计算的 TPM 可以保证所有基因的 TPM 之和在每个样本中都是一百万,因此更适合不同样本之间的直接比较。
最后总结一下
-
Counts :原始的 reads 计数值,受基因长度和测序深度影响,无法直接比较。 -
FPKM :每千碱基的转录本在每百万 reads 中的片段数,校正了基因长度和测序深度,但不适合不同样本间的比较。 -
TPM :每百万转录本中的某基因转录本数,校正了长度后标准化,使得不同样本之间的表达量可以直接比较。
在实际分析中,如果是比较不同基因的相对表达量,可以使用 FPKM;如果需要比较不同样本之间的表达情况,我们一般会选择TPM。
写作不易,欢迎关注