大家早上好,今天跟大家分享一篇今年发表在Adv Sci的一区14分的文章《Single‐Cell RNA Sequencing Identifies MMP11+ Cancer‐Associated Fibroblasts as Drivers of Angiogenesis and Bladder Cancer Progression》。该研究通过单细胞RNA测序鉴定了MMP11+成纤维细胞亚群(MMP11+ mCAFs)作为膀胱癌进展和血管生成的关键驱动因素,揭示其通过WNT5A-MCAM轴促进ESM1+ tEC尖端细胞迁移,通过CCL2/CCL11募集SPP1+巨噬细胞产生VEGFA,并受肿瘤细胞分泌的BMP2调控形成。

研究主要结果
与晚期肿瘤和不良预后相关的基因集在成纤维细胞中高表达
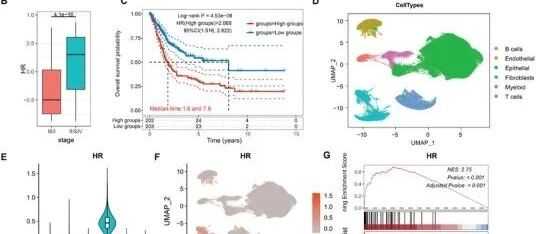
晚期肿瘤与早期肿瘤相比表现出不同的转录改变。为研究这一点,我们分析了TCGA-BLCA队列中III/IV期和I/II期肿瘤之间的差异表达基因(DEGs),鉴定出107个上调基因和18个下调基因。单变量Cox回归分析进一步显示,59个上调的DEGs与不良预后相关,而7个下调基因与良好预后相关(图1A;图S1A和表S1,补充信息)。这59个上调基因被指定为高风险(HR)基因集,在III/IV期肿瘤中显示出显著更高的表达(图1B)。Kaplan-Meier生存分析证实,HR表达升高与TCGA-BLCA队列中总生存期降低相关(HR=2.068,95% CI=1.516-2.822,p=4.53e-6)(图1C)。
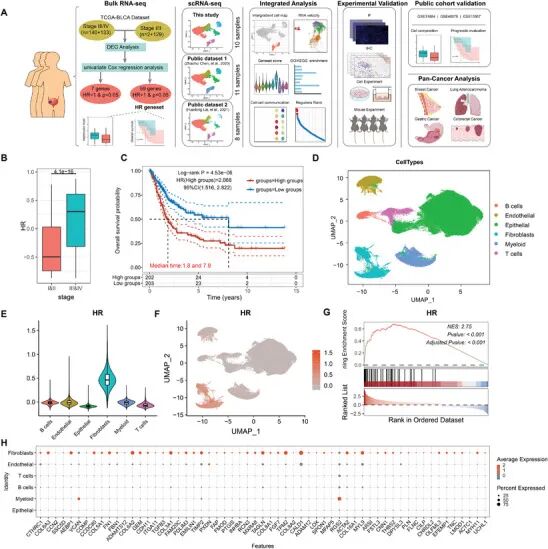
为研究HR基因集的细胞表达模式,我们构建了一个单细胞RNA测序文库,包括来自我们内部数据集的29个膀胱肿瘤和邻近正常组织,以及两个公开可用的数据集(表S2,补充信息)。经过严格的质量控制和批次校正后,184,996个细胞被聚类为29个亚群,并根据已建立的标志基因分类为六种主要细胞类型(图S1B-D,补充信息;图1D)。HR基因集特征评分显示,成纤维细胞在所有细胞类型中表现出最高的表达(图1E、F),基因集富集分析(GSEA)进一步支持了这一点,显示HR基因集在成纤维细胞中显著富集(图1G)。对单个基因表达的检查证实,大多数HR基因主要由成纤维细胞表达(图1H)。这些发现表明,成纤维细胞是晚期膀胱肿瘤中与不良预后相关的HR基因集的主要来源。
HR基因集在MMP11+成纤维细胞中高表达
为研究成纤维细胞的异质性,我们分离并重新聚类成纤维细胞群为八个不同的亚群,每个亚群根据其顶级标志基因进行标记(图2A;图S2A,补充信息)。其中,MMP11+ mCAFs表现出最高的HR特征评分(图2B),COL1A1、MMP11、POSTN、COL3A1和CTHRC1等基因表达升高(图S2B,补充信息)。比例分析显示,与邻近正常组织相比,MMP11+ mCAFs在肿瘤组织中更丰富(图2C)。使用COL1A1作为泛成纤维细胞标志物和MMP11作为特异性标志物对肿瘤和邻近正常组织切片的免疫荧光(IF)染色证实了MMP11+ mCAFs在肿瘤微环境中的主要存在,支持其潜在的功能相关性(图2D)。

为评估MMP11+ mCAFs的临床意义,我们使用CIBERSORTx对TCGA-BLCA bulk RNA-seq数据进行反卷积分析,估计每个成纤维细胞亚群的丰度。MMP11+ mCAFs的比例随着肿瘤进展逐渐增加,并主要富集在高级别肿瘤中(图2E;图S2C,补充信息)。在三个独立的GEO数据集(GSE31684、GSE48075和GSE13507)中的验证证实了类似的趋势(图2E)。重要的是,在所有四个数据集中,MMP11+ mCAFs的较高丰度始终与较差的总生存期相关(图2F)。相比之下,另一个成纤维细胞亚群IGLC1+ CAFs随着肿瘤进展显示出下降趋势,并与更好的预后相关(图S2D、E,补充信息)。值得注意的是,在所有四个数据集中,MMP11+ mCAFs和IGLC1+ CAFs之间观察到显著的负相关(图S2F,补充信息)。这些发现表明,MMP11+ mCAFs促进肿瘤进展,而IGLC1+ CAFs可能与更有利的临床结果相关。
对MMP11+ mCAFs的进一步功能分析显示,差异表达基因(DEGs)在与ECM组织、血管发育、细胞粘附、BMP和TGFβ1信号响应相关的通路中显著富集(图2G、H)。GSEA进一步证明了在标志通路如上皮-间充质转化、血管生成、凝血和顶端连接形成中的强富集。总的来说,这些发现表明MMP11+ mCAFs可能在促进肿瘤血管生成中起关键作用(图2I)。
MMP11+成纤维细胞通过WNT5A促进ESM1+尖端内皮细胞的迁移
为阐明MMP11+ mCAFs影响肿瘤微环境的机制,我们对所有细胞类型进行了全面的细胞-细胞通讯分析,确定非经典WNT(ncWNT)信号通路在MMP11+ mCAFs中特别活跃(图3A;图S3A,补充信息)。WNT5A-MCAM轴成为这种活性的主要驱动因素(图S3B,补充信息),MMP11+ mCAFs通过这一轴对内皮细胞施加强大影响(图3B)。为表征内皮细胞区室,内皮细胞被进一步分离并重新聚类为四个不同的簇,使用经典和细胞特异性标志基因:ACKR1+ vEC(静脉内皮细胞)、MALAT1+ vEC、ESM1+ tEC(尖端内皮细胞)和FBLN5+ aEC(动脉内皮细胞)(图3C;图S3C-E,补充信息)。值得注意的是,MCAM主要富集在ESM1+ tECs中(图3D),表明MMP11+ mCAFs可能优先靶向这一内皮细胞亚群。

对ESM1+ tECs的功能分析显示在与ECM重塑、血管生成、内皮细胞迁移、缺氧响应、ncWNT信号和局部粘附相关的通路中显著富集(图3E;图S3F,补充信息)。基因集变异分析(GSVA)进一步显示,ESM1+ tECs和FBLN5+ aECs都在氧化磷酸化、蛋白质分泌、脂质生成、DNA修复、PI3K-Akt-mTOR信号和TGF-β信号通路中表现出富集。重要的是,ESM1+ tECs显示出与血管生成相关程序和E2F靶标活性的更强关联,表明其在血管重塑和发育中的作用(图3F)。
为探索MMP11+ mCAFs和ESM1+ tECs之间的分子相互作用,我们应用了NicheNet分析(图S3G,补充信息)。ESM1+ tECs中的潜在靶基因富集在血管发育、内皮细胞增殖、ECM重塑和缺氧响应等生物过程中,证实了ESM1+ tECs的功能特征并支持其在血管生成中的关键作用(图3E;图S3H,补充信息)。
越来越多的证据强调WNT5A和MCAM作为血管生成的关键驱动因素。鉴于尖端内皮细胞通过定向迁移引导血管生成的作用,以及WNT5A-MCAM轴在调节细胞运动和趋同延伸中的已知功能,我们假设MMP11+ mCAFs通过这一通路促进ESM1+ tECs的迁移。肿瘤切片的IF染色显示,MMP11+ mCAFs在空间上与内皮细胞相邻,为潜在的旁分泌信号提供了解剖学证据(图3G)。Transwell迁移和管形成实验表明,WNT5A显著增强了人脐静脉内皮细胞(HUVECs)的迁移和管形成能力。相反,WNT5A抑制剂Box5-TFA损害了这两个过程,强调了WNT5A信号在内皮激活中的重要性(图3H-L)。这些结果表明,MMP11+ mCAFs可能通过WNT5A信号促进ESM1+ tECs迁移来促进血管生成。
我们进一步检查了内皮细胞中活跃的转录因子。ESM1+ tECs显示出转录因子活性升高,包括SOX18、几个HOX和ATF家族成员,以及RARG、ING4、MXD4和NFIC(图3M)。这些因子靶向ESM1+ tECs特异性的标志基因,HOXB9直接调节ESM1(一个公认的血管生成相关基因),SOX18靶向EGFL7(一个参与内皮细胞粘附、细胞骨架重塑和迁移的基因)。NFIC被发现调节FSCN1,一个对细胞迁移至关重要的肌动蛋白交联剂(图3N)。此外,先前的研究已经确定SOX18和HOXB9是关键的促血管生成因子,而SOX18、NFIC和HOXB9已被牵涉到促进细胞迁移中。
为进一步探索这些转录因子的上游调控,我们检查了WNT5A-MCAM轴与SOX18、HOXB9和NFIC表达之间的关系。观察到MCAM表达与这些转录因子之间呈正相关(图3O)。为实验验证WNT5A在转录调控中的作用,我们在体外用WNT5A及其抑制剂Box5-TFA处理HUVEC细胞,并量化SOX18、HOXB9和NFIC的表达。结果显示,WNT5A刺激显著上调了这些基因(图S3I-K,补充信息)。基于这些发现,我们推测MMP11+ mCAFs可能有助于WNT5A的上调,进而调节ESM1+ tECs中特定转录因子如SOX18、HOXB9和NFIC的表达,潜在地促进血管生成。
MMP11+成纤维细胞介导SPP1+巨噬细胞浸润和VEGFA产生
为探索MMP11+ mCAFs与肿瘤免疫微环境之间的关联,我们根据MMP11+ mCAFs的特征基因表达谱将TCGA-BLCA数据集的样本分为两个不同的分子亚型(图S4A和表S3,补充信息)。与亚型1相比,亚型2表现出显著更高的基质和免疫评分、ESTIMATE评分、MMP11+ mCAF特征评分和MMP11+ mCAFs比例,以及显著较低的肿瘤纯度(图S4B,补充信息)。免疫浸润分析显示亚型2中巨噬细胞浸润增加(图S4C,补充信息),促使进一步研究髓系细胞。
髓系细胞被分离并根据经典和簇特异性标志基因重新聚类为单核细胞、巨噬细胞、肥大细胞、cDC1、cDC2、mregDC和pDC(图S4D、E,补充信息)。值得注意的是,与正常组织相比,巨噬细胞、cDC2和mregDC在肿瘤组织中更丰富(图S4F,补充信息)。细胞-细胞通讯分析显示,MMP11+ mCAFs通过趋化因子(包括CCL11、CXCL1和CXCL6)募集髓系细胞(图S4F,补充信息),MMP11+ mCAFs与单核细胞/巨噬细胞之间最强的通讯由CXCL12、CCL11、CCL2和CCL5介导(图S4G,补充信息)。这表明MMP11+ mCAFs在肿瘤微环境中协调髓系细胞浸润。
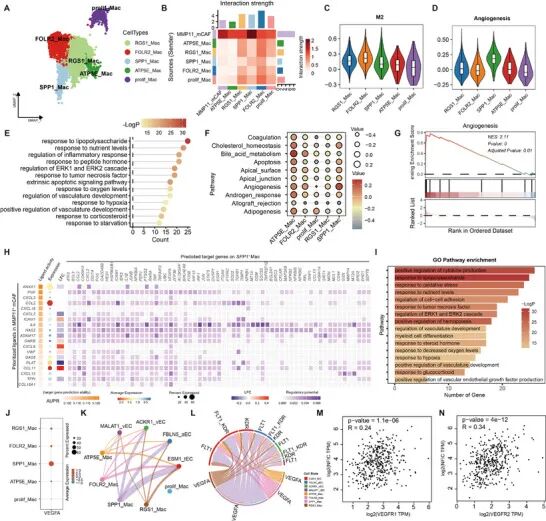
鉴于观察到的巨噬细胞浸润(图S4C,补充信息),我们进一步将巨噬细胞重新聚类为五个亚组:FOLR2+ Mac、SPP1+ Mac、RGS1+ Mac、ATP5E+ Mac和增殖Mac(图4A;图S4H,补充信息)。MMP11+ mCAFs与巨噬细胞之间最强的相互作用在SPP1+ Mac亚组中观察到(图4B)。值得注意的是,SPP1+ Mac以及RGS1+ Mac和FOLR2+ Mac经历了M2极化,SPP1+ Mac表现出最高的血管生成评分(图4C、D)。功能富集分析显示,SPP1+ Mac显著参与细胞信号传导和血管发育(图4E-G),突显其通过增强促血管生成信号在促进肿瘤血管生成中的作用。
为探索MMP11+ mCAFs和SPP1+ Mac之间的通讯,我们使用NicheNet识别由MMP11+ mCAFs在SPP1+ Mac中调控的靶基因(图4H)。该分析显示,MMP11+ mCAFs通过多个信号通路影响SPP1+ Mac,ANXA1成为细胞因子如CCL2、CXCL2、IL1B和TNF的关键调控因子。在配体中,CCL2和CCL11表达最突出(图4H)。靶基因的基因本体(GO)富集突出了它们参与细胞因子产生、营养反应、缺氧适应和血管生成(图4I)。此外,识别出与VEGFA产生正调控相关的特定GO术语,VEGFA是一种关键的血管生成介质(图4I)。NicheNet分析证实,CCL2和CCL11作为VEGFA表达的诱导剂(图4H)。表达分析显示,VEGFA主要在SPP1+ Mac中表达(图4J),这使我们假设MMP11+ mCAFs通过这些信号通路促进SPP1+ Mac中的VEGFA产生,从而促进血管生成。
巨噬细胞和内皮细胞之间的进一步通讯分析显示,主要源自SPP1+ Mac的VEGFA信号靶向ESM1+ tECs(图4K)。这种相互作用由VEGFR1和VEGFR2介导,它们调节ESM1+ tEC功能(图4L)。相关性分析显示,VEGFA信号与NFIC表达呈正相关(图4M、N)。总之,我们提出MMP11+ mCAFs募集并激活SPP1+ Mac上调VEGFA产生,进而影响ESM1+ tECs并促进血管生成。
干扰素相关基底样肿瘤细胞分泌BMP2诱导MMP11+成纤维细胞特征基因的表达
为研究MMP11+ mCAFs的起源,我们对所有成纤维细胞群进行了RNA速度分析。结果表明,MMP11+ mCAFs处于分化的起始点(图5A;图S5A-C,补充信息)。PAGA分析显示了三条分化轨迹:MMP11+ mCAFs转变为IGLC1+ CAFs、CXCL14+ CAFs,并通过PLA2G2A+ CAFs进展到PTGDS+ CAFs(图5B)。鉴于MMP11+ mCAFs随肿瘤发展逐渐增加,我们假设这一群体可能沿这些途径逆转。来自Slingshot、Cytotrace和Monocle3分析的进一步确认支持MMP11+ mCAFs代表低分化状态(图S5D-I,补充信息)。MMP11+ mCAFs的拟时序轨迹分析显示在与ECM重塑、胶原代谢、内皮细胞增殖、BMP结合和免疫系统调节相关的基因模块中显著富集(图S5J-L,补充信息)。

我们基于反映RNA速度动态的MMP11+ mCAF特征基因构建了基因集,并确定TGFB1、IL1B、BMP2和SPP1信号作为关键调节剂(图5C)。进一步研究显示,主要由上皮细胞分泌的BMP2在肿瘤微环境中调节MMP11+ mCAFs(图5D;图S7A,补充信息)。此外,MMP11+ mCAFs表现出相对较高的BMP信号受体活性(图5E、F),表明BMP2信号驱动其形成。
为探索上皮细胞中BMP2的表达,上皮细胞被重新聚类为18个不同的簇,BMP2主要在Ep3簇中表达,该簇主要来源于肿瘤(图5G、H;图S6A,补充信息)。拷贝数变异(CNV)分析证实Ep3簇主要由肿瘤细胞组成(图5I;图S6B,补充信息)。肿瘤细胞表征显示,Ep3细胞表现出干扰素相关基因(IFI27、IFI6)的高表达,并参与抗病毒免疫反应、细胞应激、免疫调节和胆固醇稳态(图S6C-F,补充信息)。进一步分析在Ep3细胞中识别出基底、应激、缺氧、pEMT、干扰素和氧化磷酸化特征(图S6G,补充信息),将它们分类为干扰素相关基底样肿瘤细胞(IFN-BL肿瘤细胞)。CellPhoneDB分析显示Ep3和MMP11+ mCAFs之间存在强相互作用(图S6H,补充信息),而CellChat分析表明来自Ep3的BMP信号主要靶向MMP11+ mCAFs(图5J)。这些发现支持IFN-BL肿瘤细胞分泌BMP2,驱动MMP11+ mCAFs形成的假设。
接下来我们分析了成纤维细胞簇内的转录因子,确定NFE2L3、RARB、TCF12和ING4为MMP11+ mCAFs中最特异的转录因子(图5K、L;图S7B、C,补充信息)。CREB3L1在活性方面排名很高(图5L)。对活性或特异性最高的前15个转录因子的分析显示,CREB3L1调控大多数MMP11+ mCAF特异性基因,而NFE2L3主要调控WNT5A(图5M)。在TCGA-BLCA样本中,NFE2L3、CREB3L1和WNT5A表达与BMP信号受体ACVR1和BMPR2呈正相关(图5N;图S7D,补充信息)。此外,在MMP11+ mCAFs中,NFE2L3活性与BMP受体表达和WNT5A相关(图S7E,补充信息)。体外定量实验表明,BMP2处理诱导了转录因子NFE2L3和CREB3L1的表达,以及MMP11+ CAFs的几个特征基因,包括WNT5A、MMP11和POSTN(图S7F-J,补充信息)。这些结果表明,IFN-BL肿瘤细胞可能通过BMP信号促进MMP11+ mCAFs中CREB3L1和NFE2L3的表达和活性,从而诱导MMP11+ mCAF特征基因的表达。
BMP2在小鼠膀胱癌模型中促进肿瘤生长和血管生成
为研究BMP2在膀胱癌进展中的作用,我们用BMP2及其小分子抑制剂Dorsomorphin处理C57BL/6J小鼠膀胱原位肿瘤模型。与PBS对照组相比,BMP2处理显著增加了肿瘤负荷,实验终点的肿瘤重量更高(图6A-D)。相反,用Dorsomorphin抑制BMP信号减轻了肿瘤进展(图6A-D),表明BMP2在体内增强肿瘤生长。
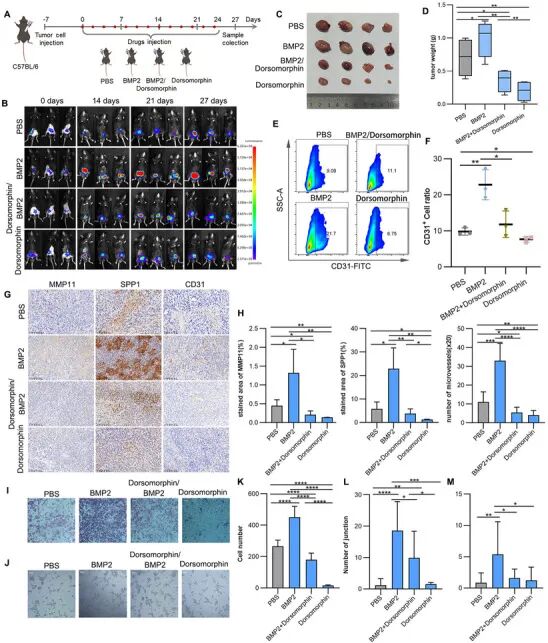
流式细胞术分析进一步显示,与对照组相比,BMP2处理组的血管内皮细胞显著增加,而Dorsomorphin处理降低了它们的丰度(图6E、F),表明BMP2促进肿瘤微环境内的血管生成。免疫组化(IHC)和IF分析显示,BMP2处理的肿瘤中MMP11+、SPP1+和CD31+细胞明显富集,而抑制剂处理组观察到相反的模式(图6G、H;图S8A、B,补充信息)。SPP1+细胞在PBS和BMP2处理组中都显示出清晰的聚集,而BMP2处理肿瘤中的CD31+内皮细胞形成了更密集和更复杂的血管结构。
鉴于BMP2可能诱导WNT5A的表达(图5M、N;图S7E、H,补充信息),已知其促进内皮细胞迁移和管形成,我们进行了条件培养基转移实验。成纤维细胞(HBdSF)用BMP2以及BMP通路抑制剂Dorsomorphin和LDN193189处理。然后将产生的条件培养基转移到HUVECs。来自BMP2处理的HBdSFs的条件培养基显著增强了HUVEC迁移和管形成,而抑制剂明显抑制了这些过程(图6I-M;图S8C-H,补充信息)。总之,我们的结果表明,BMP2通过诱导WNT5A和调节内皮细胞行为,增强小鼠膀胱癌模型中的肿瘤生长和血管生成。
MMP11+成纤维细胞在各种癌症中广泛存在
为研究MMP11+ mCAFs在各种癌症类型中的存在,我们进行了泛癌症分析。来自乳腺癌、胃癌、结直肠癌和肺腺癌的成纤维细胞进行了降维和聚类(图7A、C、E、G)。使用MMP11+ mCAFs的特征基因,我们构建了用于GSEA分析的基因集。结果显示,MMP11+ mCAFs在乳腺癌中主要与FN1+ CAFs重叠,在结直肠癌中与COL11A1+ CAFs重叠,在肺腺癌中与CTHRC1+ CAFs重叠。在胃癌中,MMP11+ mCAFs主要与COL11A+ CAFs相关,次要与CXCL5+ CAFs相关(图7B、D、F、H)。

我们进一步应用CIBERSORT反卷积算法估计这些癌症对应的TCGA数据集中MMP11+ CAFs的比例,并探索它们与肿瘤进展的关系。分析显示,MMP11+ mCAFs的比例在胃癌、乳腺癌和肺腺癌的不同阶段差异显著。在胃癌和结直肠癌中,MMP11+ mCAFs的患病率随肿瘤进展而增加,尽管结直肠癌的阶段特异性差异在统计学上不显著(图7M)。尽管存在这种变异性,生存分析表明MMP11+ mCAFs在所有四种癌症类型中始终与不良预后相关(图7I-L)。这些发现表明,虽然MMP11+ mCAFs在某些癌症中与肿瘤进展相关,但它们通常在多种恶性肿瘤中与不良临床结果相关,突显了它们作为癌症治疗中通用治疗靶点的潜力。
对这个思路感兴趣的老师可以联系。