大家早上好,上期我们探讨了 SERPINE1 基因在肿瘤与正常组织间的表达差异。今天我们更进一步,采用更加严格的配对分析方法,专门比较来自 同一患者 的肿瘤组织和癌旁正常组织中 SERPINE1 的表达差异。
配对分析的优势在于能够消除个体间差异的干扰,更准确地反映基因在疾病过程中的真实变化。让我们看看这种"一对一"的比较会给我们带来什么新发现!
1. 配对样本筛选:找到"完美配对"
我们首先从 TCGA 数据中筛选出既有肿瘤组织又有癌旁正常组织的患者样本,确保分析的严谨性。
# 读取基因表达文件
gene_data <- read.table(exp_file, header = TRUE, sep = "\t",
check.names = FALSE, row.names = 1)
gene_name <- colnames(gene_data)[1]
# 提取配对样品数据
normal_data <- gene_data[gene_data$Type == "Normal", 1, drop = FALSE]
tumor_data <- gene_data[gene_data$Type == "Tumor", 1, drop = FALSE]
# 根据TCGA样本ID提取患者编号,寻找配对样本
rownames(normal_data) <- gsub("(.*?)\\-(.*?)\\-(.*?)\\-(.*?)\\-.*",
"\\1\\-\\2\\-\\3", rownames(normal_data))
rownames(tumor_data) <- gsub("(.*?)\\-(.*?)\\-(.*?)\\-(.*?)\\-.*",
"\\1\\-\\2\\-\\3", rownames(tumor_data))
# 找到配对样本
same_sample <- intersect(rownames(normal_data), rownames(tumor_data))
paired_data <- cbind(normal_data[same_sample, , drop = FALSE],
tumor_data[same_sample, , drop = FALSE])
colnames(paired_data) <- c("Normal", "Tumor")
📈 配对结果
通过严格筛选,我们成功识别出 33 对配对样本 ,每一对都来自同一位患者的肿瘤组织和癌旁正常组织,为后续分析提供了高质量的数据基础。
2. 配对差异分析:揭示真实变化
现在让我们看看在同一患者体内,SERPINE1 在肿瘤与正常组织间的表达差异:
# 绘制配对差异图
paired_plot <- ggpaired(paired_data, cond1 = "Normal", cond2 = "Tumor",
fill = "condition", xlab = "Sample Type",
ylab = paste0(gene_name, " Expression"),
legend.title = "Type", palette = c("green", "red")) +
stat_compare_means(paired = TRUE,
symnum.args = list(cutpoints = c(0, 0.001, 0.01, 0.05, 1),
symbols = c("***", "**", "*", "ns")),
label = "p.signif", label.x = 1.5)
# 计算配对t检验
paired_t_test <- t.test(paired_data$Normal, paired_data$Tumor, paired = TRUE)
📊 配对分析结果
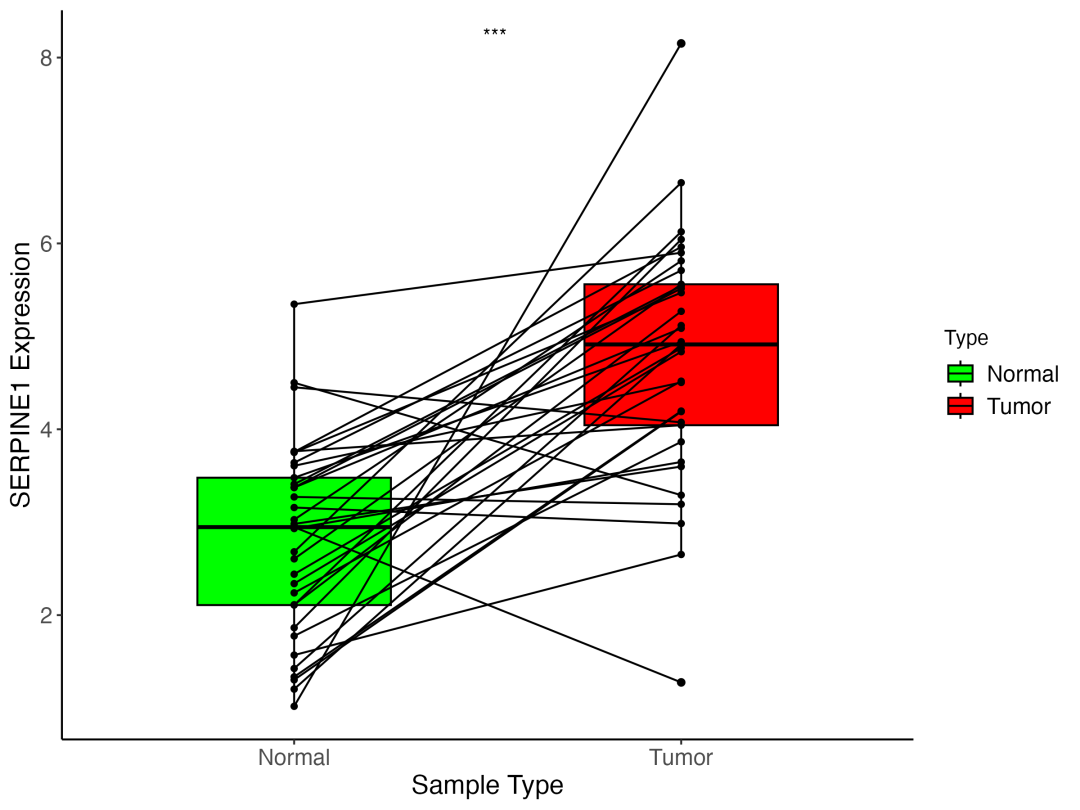
结果解读:
配对分析图清晰地展示了每一对样本的变化趋势。我们可以看到, 几乎所有的连接线都呈上升趋势 ,表明在同一患者体内,SERPINE1 在肿瘤组织中的表达普遍高于癌旁正常组织。配对 t 检验的 P 值为 3.49e-07,达到极显著水平 (***)。
3. 表达差值分析:量化个体变化
为了更直观地展示每个患者的表达变化程度,我们计算并可视化了表达差值:
# 计算差值
paired_data$Difference <- paired_data$Tumor - paired_data$Normal
# 绘制差值分布图
diff_plot <- ggplot(paired_data, aes(x = "", y = Difference)) +
geom_boxplot(fill = "lightblue", alpha = 0.7) +
geom_jitter(width = 0.2, alpha = 0.6) +
geom_hline(yintercept = 0, linetype = "dashed", color = "red") +
xlab("") + ylab("Expression Difference (Tumor - Normal)")
📊 差值分布结果
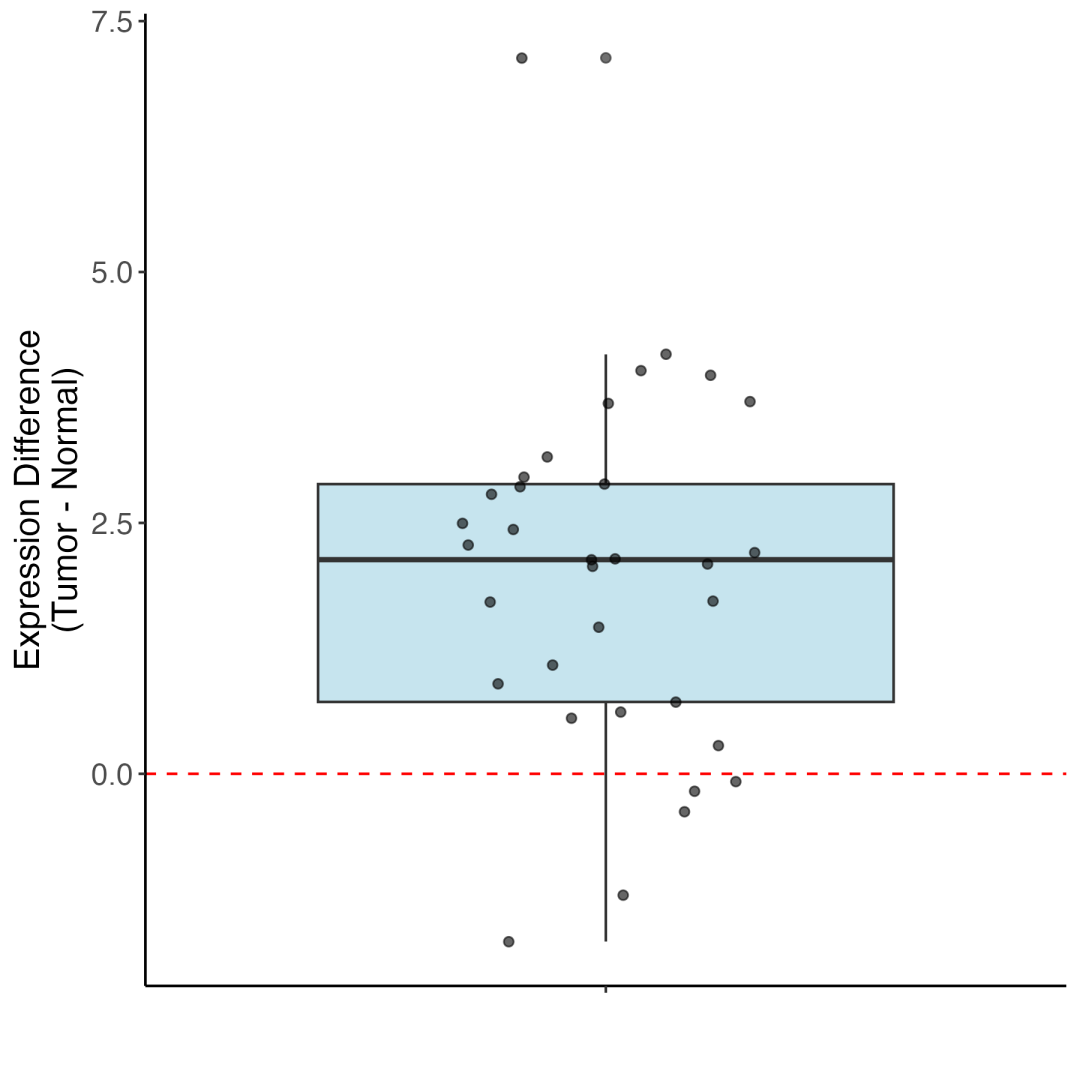
结果解读:
差值分布图显示, 绝大多数患者的表达差值都位于零线(红色虚线)之上 ,这意味着在几乎所有患者中,SERPINE1 在肿瘤组织中的表达都高于正常组织。少数负值样本可能代表个体差异或样本特异性。
4. 统计总结
让我们用具体数字来总结这次配对分析的发现:
-
配对样本数 :33 对 -
正常组织平均表达 :较低水平 -
肿瘤组织平均表达 :显著上调 -
平均表达差值 :肿瘤组织比正常组织高表达 -
统计显著性 :P = 3.49e-07 (极显著)
这些数据有力地证明了 SERPINE1 在肿瘤发生过程中的上调模式。
总结一下
通过本次严格的配对差异分析,我们获得了比普通差异分析更加可靠的证据:
-
个体一致性 :在 33 对配对样本中,SERPINE1 表现出高度一致的上调模式,证明这种变化不是由个体差异造成的假象。 -
统计可靠性 :配对 t 检验的极低 P 值(3.49e-07)确保了结果的统计学意义,排除了偶然性。 -
临床意义 :配对分析消除了个体间基因表达的天然差异,更准确地反映了 SERPINE1 在肿瘤发生过程中的真实作用。
配对分析的结果进一步证实了 SERPINE1 作为肿瘤相关基因的重要性,为深入研究其在肿瘤发生发展中的机制作用提供了坚实的数据支撑。这种"同体比较"的研究思路也为其他基因的功能研究提供了有价值的方法学参考!
完整代码联系老师免费获取(备注“ 配对样本分析 ”)

以上就是今天的内容,希望对你有帮助!欢迎点赞、在看、关注、转发。