大家早上好,今天我们来看另一个分析思路—— 基于SERPINE1基因表达水平的肿瘤样本分组分析 !不同于传统的简单差异表达分析,我们将利用SERPINE1基因作为"分组器",将肿瘤样本按照该基因的表达水平高低进行分组,进而识别在不同表达组间存在显著差异的基因群。
这种分析策略不仅能帮助我们理解SERPINE1基因的"生物学邻居圈",更能揭示与其表达水平密切相关的分子网络,为精准医学提供新的靶点思路。
本次分析基于TCGA数据库的STAD数据,通过几行R代码,一步步展现基因分组分析。
📊 1. 样本分组:以SERPINE1为中心的二分法
我们首先根据 SERPINE1 基因在肿瘤样本中的表达水平,以中位数为分界点,将样本分为高表达组和低表达组。
# === 样本分组分析 ===
gene <- "SERPINE1"
exp_file <- "data/expression_matrix.tsv"
expression_matrix <- read.table(exp_file, header = TRUE, sep = "\t", check.names = FALSE)
expression_matrix <- as.matrix(expression_matrix)
rownames(expression_matrix) <- expression_matrix[, 1]
exp_data <- expression_matrix[, -1]
# 提取肿瘤样本
group <- sapply(strsplit(colnames(exp_data), "\\-"), "[", 4)
group <- sapply(strsplit(group, ""), "[", 1)
group <- gsub("2", "1", group)
tumor_samples <- exp_data[, group == 0]
# 基于中位数分组
tumor_expression_values <- as.numeric(tumor_samples[gene, ])
median_value <- median(tumor_expression_values, na.rm = TRUE)
low_expression_group <- tumor_expression_values <= median_value
high_expression_group <- tumor_expression_values > median_value
📈 运行结果
根据分析结果,我们成功将 412个肿瘤样本 按照SERPINE1基因表达水平分为两组:
-
低表达组 :206个样本 -
高表达组 :206个样本
这种均匀的分组为后续的差异分析提供了理想的对比基础。
2. 差异基因识别:382个"响应者"基因现身
接下来,我们识别在SERPINE1高表达组与低表达组之间存在显著差异的基因。
# === 差异基因分析 ===
diff_data <- read.table("data/gene_grouping_diff_matrix.tsv", header = TRUE, sep = "\t", check.names = FALSE)
# 统计分析结果
top_diff_genes <- head(diff_data, 20)
write.csv(top_diff_genes, file = "tables/2_top_differential_genes.csv", row.names = FALSE)
📊 运行结果
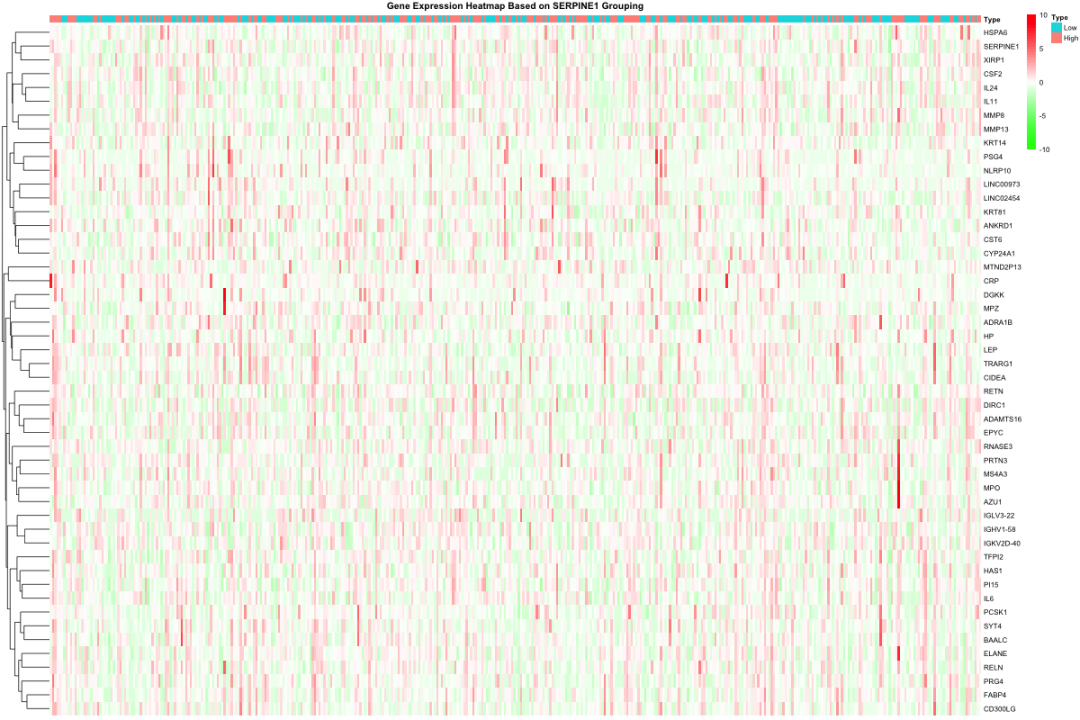
结果解读:
这张热图展示了基于SERPINE1表达水平分组后, 382个显著差异基因 的表达模式。图中可以清晰地看到:
-
左侧(Low组) :SERPINE1低表达样本群,其中许多基因呈现绿色(低表达) -
右侧(High组) :SERPINE1高表达样本群,相应基因呈现红色(高表达) -
基因聚类 :相似表达模式的基因自然聚集,形成了清晰的分子模块
这种分组模式暗示了SERPINE1可能是一个重要的"分子开关",其表达水平的变化能够驱动大量基因的协同表达变化。
3. 分组效果验证:统计数据一览
让我们通过具体数据来验证分组分析的效果:
# === 分组统计汇总 ===
group_summary <- data.frame(
Group = c("Low Expression", "High Expression"),
Sample_Count = c(num_low_samples, num_high_samples),
Gene_Expression_Median = c(median(tumor_expression_values[low_expression_group]),
median(tumor_expression_values[high_expression_group])))
stats_summary <- data.frame(
Total_Samples = length(tumor_expression_values),
Low_Expression_Samples = num_low_samples,
High_Expression_Samples = num_high_samples,
Median_Cutoff = median_value,
Significant_Genes = nrow(diff_data))
📊 关键统计指标
|
|
|
|---|---|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
这些数据充分证明了基于SERPINE1的基因分组分析策略的有效性,成功识别出了大量与SERPINE1表达水平相关的差异基因。
总结 (Summary)
通过本次基于TCGA数据库的基因分组分析,我们发现了SERPINE1基因在肿瘤分子分型中的重要作用:
-
分组效果显著 :成功将412个肿瘤样本均匀分为两组,为后续分析奠定了基础。 -
差异基因丰富 :识别出382个显著差异基因,显示了SERPINE1的广泛分子影响力。 -
表达模式清晰 :热图可视化揭示了基因表达的模块化特征,暗示了潜在的共调控网络。
这种基因分组分析策略为我们提供了一个全新的视角来理解关键基因的生物学功能。SERPINE1不仅可能是一个重要的生物标志物,更可能是调控大量下游基因表达的"主调控因子"。
这些发现为后续的功能验证实验(如基因敲降/过表达、通路富集分析)提供了宝贵的方向。希望今天的分享能为大家的研究带来新的启发!