点击卡片关注,一起学习生信分析!
1. 背景介绍
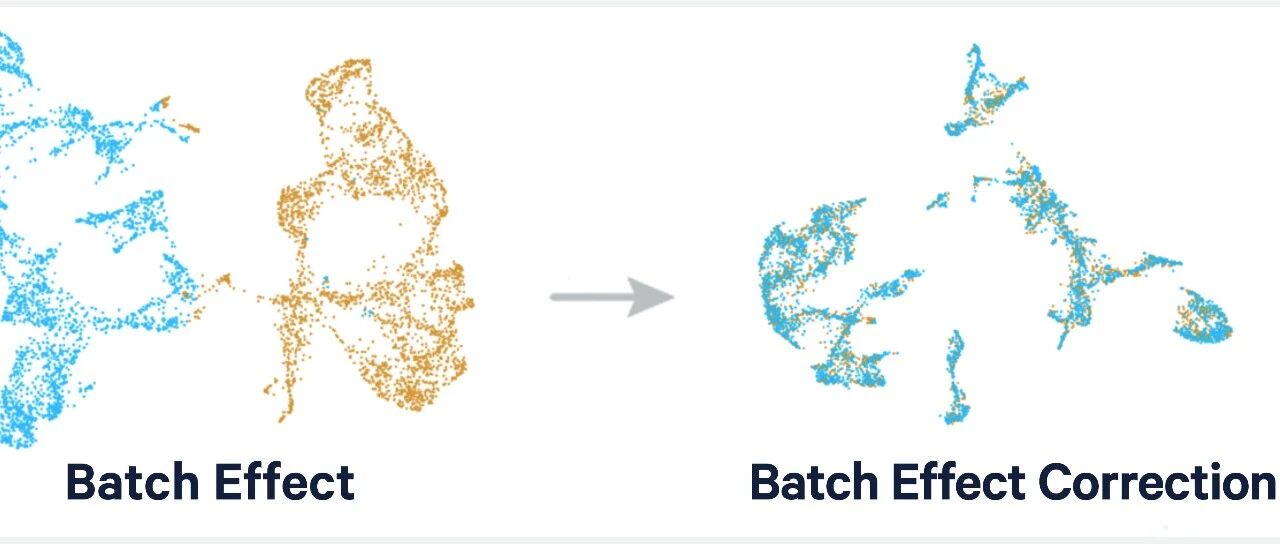
在生信分析中,我们经常需要整合来自不同来源的数据集,以增加样本量、提高统计效力或进行meta分析。然而,不同数据集之间存在批次效应(Batch Effect),即由于实验条件、测序平台、处理时间等非生物学因素导致的系统性差异。
批次效应会掩盖真实的生物学信号,导致分析结果不准确。因此,在合并多个数据集之前,必须进行批次校正(Batch Correction)。
「应用场景」 :
-
整合TCGA和GEO数据增加样本量 -
合并多个GEO数据集进行meta分析 -
消除不同测序批次间的系统性差异 -
提高差异基因检测的统计效力
2. 分析原理
2.1 批次效应的来源
「技术性批次效应」 :
-
不同的测序平台(如Illumina vs Affymetrix) -
不同的实验批次(不同时间处理的样本) -
不同的操作人员 -
不同的试剂批次
「数据类型差异」 :
-
TCGA数据:RNA-seq,TPM/FPKM/Counts -
GEO数据:芯片数据,原始信号值或标准化值
2.2 批次校正方法
本文使用 「ComBat」 方法进行批次校正:
-
ComBat是经验贝叶斯方法 -
可以有效去除批次效应 -
保留生物学变异 -
广泛应用于基因表达数据整合
2.3 数据合并流程
数据准备 → 数据标准化 → 提取共同基因 → 数据合并 → 批次校正 → 导出结果 「关键步骤」 :
-
「数据标准化」 :TCGA数据log2转换,GEO数据分位数标准化 -
「基因匹配」 :提取所有数据集的共同基因 -
「批次定义」 :标记每个样本来自哪个数据集 -
「ComBat校正」 :去除批次效应
3. 环境准备
3.1 R包安装
# 安装Bioconductor管理器
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
# 安装limma包(用于数据标准化)
BiocManager::install("limma")
# 安装sva包(包含ComBat批次校正方法)
BiocManager::install("sva")
3.2 数据准备
本教程需要三个数据集:
-
「TCGA_LUSC_TPM.txt」 :TCGA肺鳞癌数据(TPM值) -
「GSE30219.txt」 :GEO芯片数据集1 -
「GSE74777.txt」 :GEO芯片数据集2
「数据格式要求」 :
-
第一列:基因Symbol -
其余列:样本表达值 -
行:基因,列:样本
4. 代码实现
方法一:手动逐步合并(适合初学者)
这种方法逐步展示每个处理步骤,便于理解整个流程。
4.1 加载R包
# 加载必要的R包
library(limma) # 用于数据标准化
library(sva) # 用于ComBat批次校正
4.2 设置工作目录
# 设置工作目录为数据文件所在位置
setwd("你的工作目录")
# 检查当前工作目录
getwd()
# 检查数据文件是否存在
file.exists("TCGA_LUSC_TPM.txt")
file.exists("GSE30219.txt")
file.exists("GSE74777.txt")
4.3 读取数据集
# 读取TCGA数据
rt1 <- read.table("TCGA_LUSC_TPM.txt",
header = TRUE,
sep = "\t",
check.names = FALSE,
row.names = 1)
# 查看数据维度
cat("TCGA数据:", nrow(rt1), "个基因 x", ncol(rt1), "个样本\n")
# 转换为数值型矩阵
dimnames <- list(rownames(rt1), colnames(rt1))
rt1 <- matrix(as.numeric(as.matrix(rt1)),
nrow = nrow(rt1),
dimnames = dimnames)
# 查看数据前几行
rt1[1:5, 1:3]
# 读取GEO数据集1 (GSE30219)
rt2 <- read.table("GSE30219.txt",
header = TRUE,
sep = "\t",
check.names = FALSE,
row.names = 1)
dimnames <- list(rownames(rt2), colnames(rt2))
rt2 <- matrix(as.numeric(as.matrix(rt2)),
nrow = nrow(rt2),
dimnames = dimnames)
cat("GSE30219数据:", nrow(rt2), "个基因 x", ncol(rt2), "个样本\n")
# 读取GEO数据集2 (GSE74777)
rt3 <- read.table("GSE74777.txt",
header = TRUE,
sep = "\t",
check.names = FALSE,
row.names = 1)
dimnames <- list(rownames(rt3), colnames(rt3))
rt3 <- matrix(as.numeric(as.matrix(rt3)),
nrow = nrow(rt3),
dimnames = dimnames)
cat("GSE74777数据:", nrow(rt3), "个基因 x", ncol(rt3), "个样本\n")
4.4 数据标准化
不同来源的数据需要不同的标准化方法。
# 对TCGA数据进行log2转换
# TCGA的TPM数据通常未经log转换
rt1 <- log2(rt1 + 1)
cat("✓ TCGA数据log2转换完成\n")
# 对GEO芯片数据进行分位数标准化
# 使用limma包的normalizeBetweenArrays函数
rt2 <- normalizeBetweenArrays(rt2)
cat("✓ GSE30219数据标准化完成\n")
rt3 <- normalizeBetweenArrays(rt3)
cat("✓ GSE74777数据标准化完成\n")
「为什么不同数据需要不同处理?」
-
TCGA的TPM数据:线性尺度,需要log转换使其接近正态分布 -
GEO芯片数据:可能已经log转换,使用分位数标准化使样本间分布一致
4.5 提取共同基因
只有所有数据集都包含的基因才能用于合并。
# 获取前两个数据集的共同基因
gene1 <- intersect(rownames(rt1), rownames(rt2))
cat("TCGA和GSE30219的共同基因:", length(gene1), "个\n")
# 获取三个数据集的共同基因
gene2 <- intersect(gene1, rownames(rt3))
cat("三个数据集的共同基因:", length(gene2), "个\n")
# 查看共同基因示例
head(gene2, 10)
4.6 按共同基因合并数据
# 创建空数据框用于合并
allmerge1 <- data.frame()
# 合并TCGA和GSE30219
allmerge1 <- cbind(rt1[gene2, ], rt2[gene2, ])
cat("合并TCGA和GSE30219后:", nrow(allmerge1), "个基因 x", ncol(allmerge1), "个样本\n")
# 继续合并GSE74777
allmerge1 <- cbind(allmerge1, rt3[gene2, ])
cat("最终合并矩阵:", nrow(allmerge1), "个基因 x", ncol(allmerge1), "个样本\n")
# 查看合并后的数据
allmerge1[1:5, c(1:3, (ncol(rt1)+1):(ncol(rt1)+3), (ncol(allmerge1)-2):ncol(allmerge1))]
4.7 定义批次信息
ComBat需要知道每个样本属于哪个批次(数据集)。
# 创建批次标签向量
batchType1 <- c()
# TCGA数据标记为批次1
batchType1 <- c(batchType1, rep(1, ncol(rt1)))
# GSE30219标记为批次2
batchType1 <- c(batchType1, rep(2, ncol(rt2)))
# GSE74777标记为批次3
batchType1 <- c(batchType1, rep(3, ncol(rt3)))
# 查看批次信息
cat("批次信息:\n")
cat("批次1 (TCGA):", sum(batchType1 == 1), "个样本\n")
cat("批次2 (GSE30219):", sum(batchType1 == 2), "个样本\n")
cat("批次3 (GSE74777):", sum(batchType1 == 3), "个样本\n")
4.8 执行ComBat批次校正
# 使用ComBat进行批次校正
cat("\n开始ComBat批次校正...\n")
outTab1 <- ComBat(dat = allmerge1,
batch = batchType1,
par.prior = TRUE) # 使用参数先验
cat("✓ 批次校正完成\n")
「ComBat参数说明」 :
-
dat:表达矩阵(基因×样本) -
batch:批次标签向量 -
par.prior:是否使用参数先验(TRUE更稳健)
4.9 格式化并导出数据
# 在第一行添加样本ID
outTab1 <- rbind(ID = colnames(outTab1), outTab1)
# 查看校正后的数据
outTab1[1:6, 1:5]
# 导出批次校正后的数据
write.table(outTab1,
file = "merge1.txt",
sep = "\t",
quote = FALSE,
col.names = FALSE)
cat("✓ 批次校正后的数据已保存到 merge1.txt\n")
cat("最终矩阵:", nrow(outTab1)-1, "个基因 x", ncol(outTab1), "个样本\n")
方法二:循环批量处理(适合多数据集)
当需要合并多个数据集时,使用循环可以使代码更简洁高效。
4.10 准备文件列表
# 加载R包
library(limma)
library(sva)
# 设置工作目录
setwd("你的工作目录")
# 定义要合并的文件列表
files <- c("TCGA_LUSC_TPM.txt", "GSE30219.txt", "GSE74777.txt")
cat("准备合并", length(files), "个数据集\n")
for (i in 1:length(files)) {
cat(i, ".", files[i], "\n")
}
4.11 循环读取并标准化数据
# 创建列表存储每个数据集
geneList <- list()
cat("\n开始读取和处理数据...\n")
for (i in1:length(files)) {
# 读取数据
rt <- read.table(files[i],
header = TRUE,
sep = "\t",
check.names = FALSE,
row.names = 1)
# 转换为数值矩阵
dimnames <- list(rownames(rt), colnames(rt))
rt <- matrix(as.numeric(as.matrix(rt)),
nrow = nrow(rt),
dimnames = dimnames)
# 根据样本ID前缀判断数据类型并标准化
# TCGA样本ID以"TCGA"开头
if (substr(colnames(rt)[1], 1, 3) == "TCG") {
rt <- log2(rt + 1)
cat(i, ". ", files[i], " - TCGA数据,已log2转换\n")
}
# GEO样本ID以"GSM"开头
if (substr(colnames(rt)[1], 1, 3) == "GSM") {
rt <- normalizeBetweenArrays(rt)
cat(i, ". ", files[i], " - GEO数据,已标准化\n")
}
# 保存到列表
geneList[[i]] <- rt
# 获取共同基因
if (i == 1) {
gene3 <- rownames(geneList[[i]])
} else {
gene3 <- intersect(gene3, rownames(geneList[[i]]))
}
}
cat("\n所有数据集的共同基因:", length(gene3), "个\n")
4.12 循环合并数据并定义批次
# 初始化合并矩阵和批次向量
allmerge2 <- data.frame()
batchType2 <- c()
cat("\n开始合并数据集...\n")
for (i in1:length(files)) {
# 合并数据(只保留共同基因)
if (i == 1) {
allmerge2 <- geneList[[i]][gene3, ]
cat("初始化矩阵:", nrow(allmerge2), "基因 x", ncol(allmerge2), "样本\n")
} else {
allmerge2 <- cbind(allmerge2, geneList[[i]][gene3, ])
cat("合并第", i, "个数据集,当前:", nrow(allmerge2), "基因 x", ncol(allmerge2), "样本\n")
}
# 添加批次信息
batchType2 <- c(batchType2, rep(i, ncol(geneList[[i]])))
}
cat("\n合并完成!\n")
cat("批次分布:\n")
for (i in1:length(files)) {
cat("批次", i, "(", files[i], "):", sum(batchType2 == i), "个样本\n")
}
4.13 执行批次校正并导出
# ComBat批次校正
cat("\n开始ComBat批次校正...\n")
outTab2 <- ComBat(dat = allmerge2,
batch = batchType2,
par.prior = TRUE)
cat("✓ 批次校正完成\n")
# 添加样本ID行并导出
outTab2 <- rbind(ID = colnames(outTab2), outTab2)
write.table(outTab2,
file = "merge2.txt",
sep = "\t",
quote = FALSE,
col.names = FALSE)
cat("✓ 数据已保存到 merge2.txt\n")
cat("最终矩阵:", nrow(outTab2)-1, "个基因 x", ncol(outTab2), "个样本\n")
5. 结果解读
5.1 输出文件说明
运行脚本后会生成两个文件(两种方法结果相同):
「merge1.txt / merge2.txt」 :
-
第一行:样本ID -
第一列:基因Symbol -
数值:批次校正后的标准化表达值
5.2 批次校正前后对比
「校正前」 :
-
来自不同数据集的样本会聚类在一起 -
批次效应掩盖了真实的生物学分组 -
PCA图显示样本按数据集分开
「校正后」 :
-
批次效应被去除 -
样本按生物学特征(如肿瘤vs正常)分组 -
可以进行联合差异分析
5.3 数据质量检查
# 读取批次校正后的数据
merged_data <- read.table("merge1.txt", header = TRUE, sep = "\t",
check.names = FALSE, row.names = 1)
merged_data <- merged_data[-1, ] # 删除ID行
merged_data <- as.matrix(merged_data)
# 转换为数值型
mode(merged_data) <- "numeric"
# 查看数据分布
summary(as.vector(merged_data))
# 检查缺失值
sum(is.na(merged_data))
# 查看数据维度
dim(merged_data)
5.4 批次校正效果评估
「方法一:PCA分析」
# 需要安装FactoMineR包
# install.packages("FactoMineR")
library(FactoMineR)
# 进行PCA分析
pca_result <- PCA(t(merged_data), graph = FALSE)
# 可视化PCA结果
plot(pca_result, choix = "ind")
「方法二:样本相关性热图」
# 计算样本间相关性
cor_matrix <- cor(merged_data)
# 绘制热图
library(pheatmap)
pheatmap(cor_matrix,
show_rownames = FALSE,
show_colnames = FALSE)
5.5 合并数据的典型统计
假设合并了TCGA、GSE30219、GSE74777三个数据集:
|
|
|
|
|
|---|---|---|---|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 「合并后」 |
|
|
|
6. 常见问题
6.1 共同基因太少怎么办?
「问题」 :三个数据集合并后只剩下很少的基因
「原因」 :
-
GEO芯片平台不同,覆盖的基因不同 -
基因命名不一致(Symbol vs Ensembl ID)
「解决方法」 :
# 检查各数据集的基因数量
cat("TCGA基因数:", length(rownames(rt1)), "\n")
cat("GSE30219基因数:", length(rownames(rt2)), "\n")
cat("GSE74777基因数:", length(rownames(rt3)), "\n")
# 两两比较共同基因数
length(intersect(rownames(rt1), rownames(rt2)))
length(intersect(rownames(rt1), rownames(rt3)))
length(intersect(rownames(rt2), rownames(rt3)))
6.2 ComBat运行报错
「错误1」
:
batch has too many levels
「原因」 :批次数量过多,样本量太少
「解决」 :
-
确保每个批次至少有3-5个样本 -
合并相似的批次
「错误2」
:
missing values in data
「原因」 :数据中有NA值
「解决」 :
# 删除包含NA的基因
merged_data <- merged_data[complete.cases(merged_data), ]
# 或者用均值填充
merged_data[is.na(merged_data)] <- mean(merged_data, na.rm = TRUE)
6.3 数据尺度不一致
「问题」 :TCGA和GEO数据的数值范围差异很大
「检查」 :
# 查看数据范围
range(rt1, na.rm = TRUE)
range(rt2, na.rm = TRUE)
range(rt3, na.rm = TRUE)
「解决」 :确保所有数据都经过适当的标准化
-
TCGA:log2(TPM + 1) -
GEO:normalizeBetweenArrays()
6.4 如何判断是否需要批次校正?
「方法1:查看样本来源是否聚类」
# 绘制聚类热图
pheatmap(cor(allmerge1),
annotation_col = data.frame(
Batch = factor(batchType1)
))
「方法2:PCA分析」
-
如果PCA图中样本按数据集分组,需要批次校正 -
如果按生物学分组,可能不需要
6.5 批次校正过度怎么办?
过度校正可能去除真实的生物学差异。
「预防措施」 :
-
保留原始数据用于比较 -
如果有分组信息,在ComBat中使用mod参数保护生物学差异
# 创建模型矩阵保护分组信息
# group <- c(rep("Tumor", n_tumor), rep("Normal", n_normal))
# mod <- model.matrix(~as.factor(group))
# outTab <- ComBat(allmerge1, batchType1, mod = mod)
6.6 大数据集内存不足
「问题」 :合并大量样本时内存溢出
「解决」 :
# 增加R的内存限制
memory.limit(size = 50000) # Windows
# options(java.parameters = "-Xmx8g") # 增加Java堆内存
# 或者分批处理
# 先合并一部分数据集,再合并其他数据集
如果需要文章代码,联系老师免费获取。
( 请发送文章链接告知老师获取哪个代码)
送你一份科研小礼物,开启科研生涯!
点击即可领取: